Thyroid Cancer
- Overview
- Epidemiology
- Types of Thyroid Cancer
- Pathophysiology
- Risk Factors
- Clinical Presentation
- Diagnosis and Staging
- Treatment
- Prognosis
- Research Papers
- Connections
- Featured Videos
Overview
Thyroid cancer is the most common malignancy of the endocrine system, arising from the cellular components of the thyroid gland — a butterfly-shaped organ located at the base of the neck that wraps around the trachea. The thyroid produces two main hormones, thyroxine (T4) and triiodothyronine (T3), which regulate metabolism, heart rate, body temperature, and growth and development throughout life. A third hormone, calcitonin, is produced by specialized parafollicular C-cells within the gland and plays a role in calcium homeostasis.
Unlike many solid tumors, the majority of thyroid cancers carry an excellent prognosis. Differentiated thyroid cancers — papillary and follicular subtypes — retain many functional characteristics of normal thyroid tissue, including the ability to absorb iodine, which makes them responsive to radioiodine (RAI) therapy. For most patients diagnosed with differentiated thyroid cancer, surgery followed by appropriate adjuvant treatment results in long-term disease-free survival exceeding 95 percent at ten years.
However, thyroid cancer is not a single disease. The rare anaplastic subtype (less than 1 percent of cases) represents one of the most aggressive solid tumors in all of oncology, with a median survival measured in months. Understanding the subtype is therefore critical — it determines prognosis, treatment, and urgency far more than a simple “thyroid cancer” diagnosis suggests. This heterogeneity has driven intense research into molecular drivers, targeted therapies, and refined risk stratification tools that now guide individualized care.
Thyroid cancer is also notable for being one of the few cancers with a sharply rising incidence over the past three decades — a trend driven partly by increased ultrasound surveillance detecting small, subclinical nodules, and partly by a genuine rise in cancer occurrence. This overdiagnosis debate has real clinical consequences, informing contemporary guidelines that favor active surveillance over surgery for very low-risk microcarcinomas in carefully selected patients.
Epidemiology
Thyroid cancer is diagnosed in approximately 44,000 Americans each year, making it the fifth most common cancer in women and one of the fastest-growing cancer diagnoses by incidence in the United States. Worldwide, it accounts for roughly 586,000 new cases annually, with significant geographic variation linked to differences in iodine intake, radiation exposure history, and diagnostic intensity.
A striking epidemiologic feature is the pronounced female predominance: women are diagnosed approximately three times more often than men. This ratio is most marked during the reproductive years (ages 20–55), suggesting a role for estrogen or reproductive hormones in tumor initiation or growth, though the exact mechanism remains incompletely understood. After menopause, the sex disparity narrows considerably.
Incidence has risen dramatically since the 1990s — roughly tripling over three decades in some U.S. data sets. Much of this rise reflects the detection of small papillary microcarcinomas (≤1 cm) that would previously have remained undetected, discovered incidentally on neck ultrasound performed for other reasons such as carotid artery evaluation or parathyroid imaging. However, careful epidemiologic analyses have also identified a genuine increase in larger tumors (>2 cm), suggesting the trend is not entirely an artifact of surveillance. Mortality from thyroid cancer has remained relatively stable, reinforcing the view that widespread detection of indolent microcarcinomas accounts for much of the incidence rise.
Age at diagnosis varies by subtype. Papillary thyroid cancer peaks in the third to fifth decades, follicular cancer in the fourth to sixth, and anaplastic cancer almost exclusively in patients over 60. Medullary thyroid cancer occurs across a wide age range, with hereditary forms often presenting in younger patients due to genetic testing programs that identify RET mutation carriers before symptomatic disease develops.
Racial and ethnic disparities exist: Asian American and Pacific Islander populations have higher incidence rates, potentially related to differences in iodine intake and diagnostic access. Black patients are diagnosed at later stages on average and experience modestly worse outcomes despite similar tumor biology, reflecting systemic healthcare access inequities.
Types of Thyroid Cancer
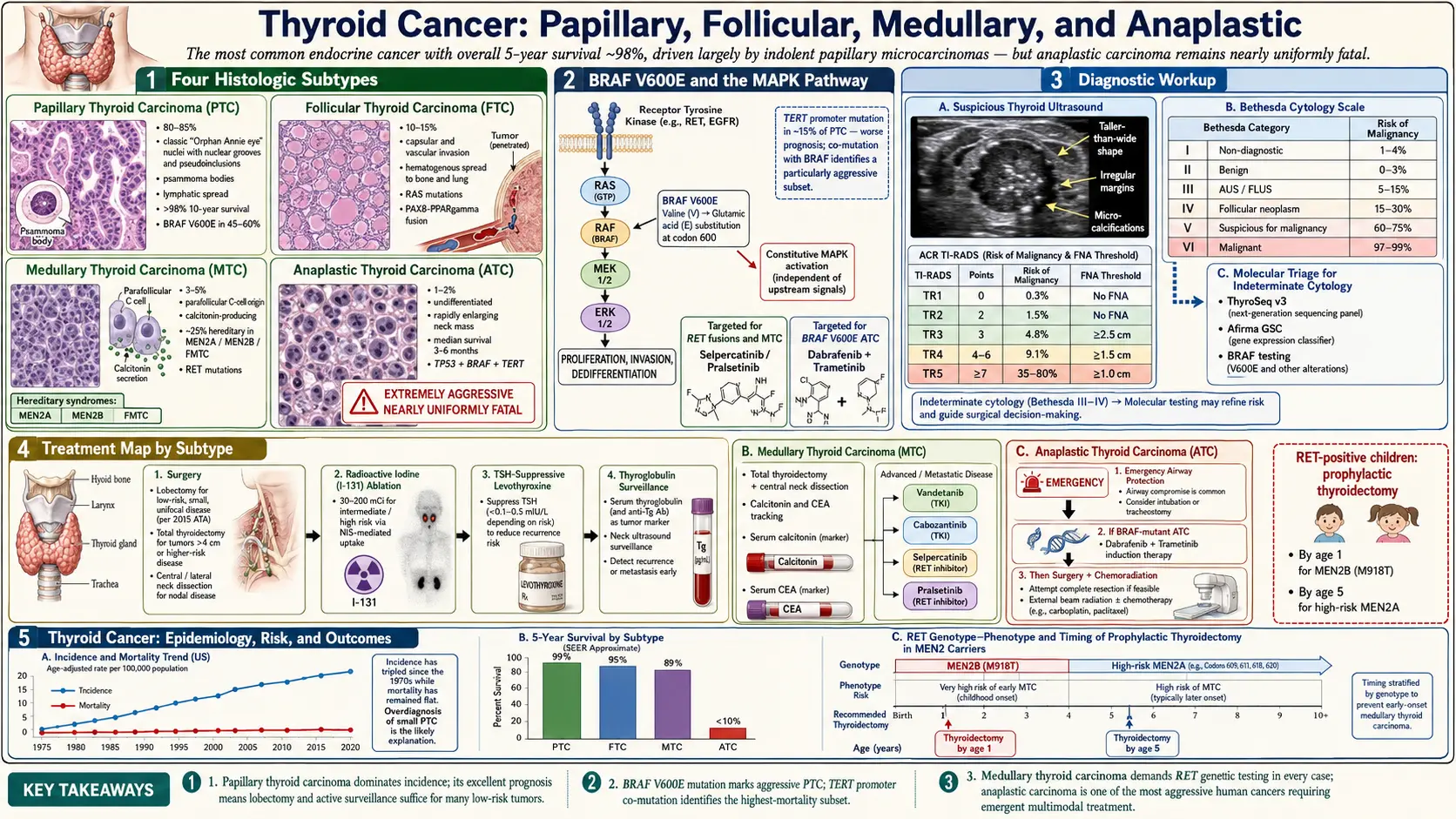
Four major histologic subtypes account for virtually all thyroid cancers, each with distinct cell of origin, molecular drivers, clinical behavior, and treatment approach.
Papillary Thyroid Cancer (PTC)
Papillary thyroid cancer is by far the most common subtype, representing approximately 85 percent of all thyroid malignancies. It arises from thyroid follicular cells and is named for its characteristic papillary architecture on pathology — finger-like projections lined by cells with distinctive nuclear features including ground-glass (“Orphan Annie eye”) nuclei, nuclear grooves, and intranuclear pseudoinclusions. PTC has a marked tendency to spread to regional cervical lymph nodes, occurring in 30–80 percent of cases depending on tumor size and variant, yet this lymph node involvement paradoxically carries limited impact on long-term survival in most patients.
The dominant molecular driver of PTC is activation of the MAPK signaling pathway. The BRAF V600E point mutation is present in approximately 45–60 percent of PTCs, substituting glutamic acid for valine at codon 600 of the BRAF kinase, producing constitutive ERK pathway activation. RET/PTC rearrangements (fusions of the RET receptor tyrosine kinase with partner genes such as CCDC6 and NCOA4) are found in 10–20 percent of PTCs, particularly in radiation-associated tumors. RAS mutations (NRAS, HRAS, KRAS) are more common in the follicular variant of PTC. These molecular alterations are the targets of contemporary targeted therapies in advanced disease and form the basis of commercial molecular diagnostic panels used to evaluate indeterminate thyroid nodules.
Several PTC variants carry distinct clinical implications. Tall cell variant and hobnail variant are associated with more aggressive behavior, higher recurrence rates, and greater BRAF mutation prevalence. The encapsulated follicular variant (EFVPTC) — now reclassified as “noninvasive follicular thyroid neoplasm with papillary-like nuclear features” (NIFTP) when non-invasive — has an extremely low malignant potential and does not require RAI or TSH suppression after lobectomy.
Follicular Thyroid Cancer (FTC)
Follicular thyroid cancer accounts for approximately 10 percent of cases. Like PTC, it arises from follicular cells, but it lacks the nuclear features that define PTC and cannot be diagnosed on fine-needle aspiration cytology alone — the distinction from follicular adenoma requires histologic demonstration of capsular or vascular invasion on the surgical specimen. This biology drives the clinical challenge of the indeterminate thyroid nodule, where molecular testing has become increasingly important.
FTC disseminates primarily by hematogenous spread to distant sites — the lungs and bones — rather than lymph nodes. Bone metastases from FTC are often lytic and can be destructive. The most common molecular alterations are RAS mutations (present in 40–50% of FTCs) and the PAX8-PPARG gene fusion (20–35%), which results from a chromosomal translocation and produces a fusion protein that may act as a dominant negative inhibitor of normal PPARG tumor suppressor function.
Minimally invasive FTC (capsular invasion only, no vascular invasion) carries an excellent prognosis comparable to PTC. Widely invasive FTC with extensive vascular invasion has a substantially worse outcome, with 10-year survival rates approaching 50 percent in some series, and is treated more aggressively with total thyroidectomy, RAI, and TSH suppression.
Medullary Thyroid Cancer (MTC)
Medullary thyroid cancer arises from the parafollicular C-cells of the thyroid, which are neural crest-derived cells that produce calcitonin. MTC accounts for approximately 5 percent of thyroid cancers. Unlike the differentiated subtypes, MTC does not take up radioiodine (C-cells lack the sodium-iodide symporter) and does not produce thyroglobulin — instead, serum calcitonin and carcinoembryonic antigen (CEA) serve as sensitive biomarkers for tumor burden, response to treatment, and surveillance for recurrence.
Approximately 75 percent of MTCs are sporadic, arising without an identifiable germline mutation. The remaining 25 percent are hereditary, caused by activating germline mutations in the RET proto-oncogene, and are classified into three syndromes: Multiple Endocrine Neoplasia type 2A (MEN2A, associated with pheochromocytoma and primary hyperparathyroidism), MEN2B (associated with pheochromocytoma, mucosal neuromas, and marfanoid habitus), and Familial Medullary Thyroid Cancer (FMTC). Specific RET codon mutations correlate with disease aggressiveness, and prophylactic thyroidectomy is recommended in RET carriers at an age determined by mutation-specific risk level — as early as the first months of life for the most aggressive MEN2B mutations at codon 918.
Even sporadic MTCs carry somatic RET mutations in approximately 40–50 percent of cases, along with somatic RAS mutations in another 15 percent. The identification of RET as the central driver has led to the development of potent, selective RET kinase inhibitors that have transformed systemic treatment of advanced MTC.
Anaplastic Thyroid Cancer (ATC)
Anaplastic thyroid cancer represents less than 1 percent of thyroid cancers but accounts for 14–39 percent of thyroid cancer deaths due to its extreme aggressiveness. ATC is thought to arise from dedifferentiation of pre-existing differentiated or poorly differentiated thyroid cancer — many ATCs contain areas of coexisting PTC or FTC — though de novo origin from follicular cells also occurs. It presents predominantly in elderly patients (median age ~70) as a rapidly growing neck mass that invades adjacent structures within weeks.
The molecular profile of ATC is complex. BRAF V600E mutations are found in approximately 45 percent of ATCs — a critical therapeutic target. TP53 mutations, which are rare in DTC, are present in 50–80 percent of ATCs and likely represent a driver of the dedifferentiation process. TERT promoter mutations, which extend telomere length and permit cellular immortality, are present in up to 75 percent of ATCs. PIK3CA, PTEN, and RAS mutations are also common, reflecting activation of both the MAPK and PI3K/AKT/mTOR pathways. The complexity of the ATC genomic landscape explains both its resistance to conventional therapy and the emerging rationale for combination targeted approaches.
By AJCC staging convention, all ATCs are classified as Stage IV at diagnosis regardless of extent — reflecting the uniformly poor prognosis. Stage IVA disease is confined to the thyroid (resectable), IVB involves extrathyroidal extension without distant metastasis, and IVC indicates distant metastasis. Multimodality treatment including surgery when feasible, external beam radiotherapy, and systemic therapy is required, but even with optimal management median overall survival is 3–5 months.
Pathophysiology
The molecular biology of thyroid cancer centers on two major oncogenic pathways: the MAPK (RAS-RAF-MEK-ERK) pathway and the PI3K/AKT/mTOR pathway. Both are downstream of receptor tyrosine kinases including RET, VEGFR, and EGFR, and both regulate proliferation, differentiation, and survival. In differentiated thyroid cancers, one of these pathways is typically activated by a single driver mutation (BRAF V600E, RET/PTC fusion, or RAS mutation), whereas anaplastic cancers accumulate multiple cooperating mutations that collectively drive the undifferentiated, highly proliferative phenotype.
A key functional property of differentiated thyroid cancer is the preserved, though reduced, expression of the sodium-iodide symporter (NIS) — the membrane transporter that actively concentrates iodide within thyroid cells. NIS expression is regulated by TSH (thyroid-stimulating hormone) signaling through the cAMP pathway. This property underpins two of the most important tools in DTC management: radioiodine (I-131) therapy, which exploits NIS to deliver lethal beta-radiation selectively to thyroid cells, and TSH suppression therapy, where supraphysiologic doses of levothyroxine suppress pituitary TSH secretion, removing a growth stimulus for residual thyroid tissue and tumor cells.
Thyroglobulin (Tg) — the large glycoprotein scaffold on which thyroid hormones are synthesized and stored — serves as a sensitive and specific serum tumor marker for differentiated thyroid cancer following total thyroidectomy. After complete surgical resection and RAI ablation of residual normal thyroid tissue, detectable or rising serum Tg indicates recurrent or persistent disease. Anti-thyroglobulin antibodies (TgAb), present in approximately 20–25 percent of DTC patients, can interfere with Tg immunoassays, requiring monitoring of TgAb trends as a surrogate marker.
BRAF V600E in PTC promotes sustained MAPK activation that downregulates NIS expression and thyroid differentiation genes (TTF-1, PAX8, thyroglobulin), contributing to RAI resistance in aggressive BRAF-mutant tumors. This has led to the concept of “redifferentiation therapy” — using MEK or BRAF inhibitors to restore NIS expression and RAI uptake in tumors that have become RAI-refractory, a strategy now being tested in clinical trials with early promising results.
In MTC, calcitonin secretion by C-cells provides a uniquely sensitive biomarker. Stimulated calcitonin testing with pentagastrin or calcium infusion can detect very small tumors, and the calcitonin doubling time during surveillance predicts survival — a doubling time under 6 months confers a 5-year survival of only 25 percent, while a doubling time over 2 years is associated with near-normal survival. CEA co-secretion by MTC cells reflects dedifferentiation and independently predicts prognosis.
Risk Factors
Several environmental, hormonal, and genetic factors influence thyroid cancer risk, though the majority of patients have no identifiable predisposing condition.
Ionizing radiation: Exposure to ionizing radiation, particularly during childhood and adolescence when the thyroid is most radiosensitive, is the strongest established environmental risk factor. External beam radiation to the head and neck region — historically used to treat acne, enlarged tonsils, and thymic enlargement — substantially increases lifetime PTC risk, with a latency period of 5–30 years. The Chernobyl nuclear accident in 1986 produced a dramatic rise in childhood PTC in exposed populations, predominantly papillary cancers driven by RET/PTC rearrangements. Diagnostic medical radiation (dental X-rays, CT scans) contributes minimally to population-level risk, though cumulative CT exposure remains a topic of ongoing study.
Iodine status: Iodine deficiency is associated with follicular and anaplastic thyroid cancer, while iodine sufficiency after correction of deficiency may increase the relative prevalence of papillary cancer. The relationship is complex and may reflect shifts in underlying thyroid pathology (goiter, nodularity) rather than direct carcinogenic effects. Excessive iodine intake is not consistently linked to thyroid cancer risk in population studies.
Female sex and hormonal factors: The 3:1 female predominance strongly implicates sex hormones, though oral contraceptive use and hormone replacement therapy have not been consistently associated with thyroid cancer in epidemiologic studies. Pregnancy is not associated with de novo development but may stimulate growth of pre-existing thyroid cancer.
Family history and genetics: Non-medullary thyroid cancer is familial in approximately 5 percent of cases (defined as 2 or more first-degree relatives with PTC or FTC), though most cases represent polygenic risk rather than single-gene inheritance. Specific cancer predisposition syndromes associated with thyroid cancer include Cowden syndrome (PTEN hamartoma tumor syndrome, FTC/PTC risk), familial adenomatous polyposis/Gardner syndrome (increased PTC risk, particularly the cribriform-morular variant), Werner syndrome, and Carney complex.
RET germline mutations: Virtually all hereditary MTC is caused by autosomal dominant activating mutations in the RET proto-oncogene. Genetic testing of all newly diagnosed MTC patients for germline RET mutations is standard of care, with cascade family testing of first-degree relatives upon identification of a pathogenic variant. Prophylactic thyroidectomy based on mutation-specific risk stratification can prevent MTC in gene carriers.
Benign thyroid disease: The presence of thyroid nodules, goiter, and Hashimoto’s thyroiditis has been associated with modestly increased thyroid cancer risk in epidemiologic studies, though the absolute risk remains low and the relationship with Hashimoto’s disease specifically is debated and may partly reflect detection bias.
Obesity and metabolic syndrome: Emerging evidence suggests associations between obesity, elevated BMI, insulin resistance, and thyroid cancer risk, possibly mediated through elevated insulin-like growth factor (IGF-1) signaling, but the data remain insufficient to alter clinical screening recommendations.
Clinical Presentation
The most common presentation of thyroid cancer is a painless, palpable thyroid nodule discovered by the patient, a family member, or a clinician on routine physical examination. Thyroid nodules are extremely common — present in up to 68 percent of adults on high-resolution ultrasound — and the vast majority are benign. Fewer than 10 percent of detected nodules prove malignant on pathologic examination, with the proportion varying based on nodule characteristics and patient risk factors.
Incidental detection: With the widespread use of neck imaging for other indications (carotid Doppler, cervical spine MRI, CT for trauma or pulmonary disease, PET-CT for other malignancies), an increasing proportion of thyroid cancers are detected as incidental findings on imaging not ordered to evaluate the thyroid. These “incidentalomas” are frequently small and low-risk, accounting for much of the rise in thyroid cancer incidence.
Cervical lymphadenopathy: Painless, firm, non-tender enlarged cervical lymph nodes may be the presenting symptom, particularly in patients with papillary cancer that has spread to regional nodes. In young patients with a lateral neck mass, metastatic PTC must be considered even if the primary thyroid lesion is small or impalpable.
Local invasion symptoms: Symptoms suggesting extrathyroidal extension indicate locally advanced disease and require urgent evaluation. Hoarseness due to recurrent laryngeal nerve (RLN) involvement is a particularly concerning sign suggesting invasive cancer, as the RLN runs in close proximity to the thyroid in the tracheoesophageal groove. Dysphagia reflects esophageal compression or invasion. Stridor indicates tracheal compression or invasion. Neck pain or dyspnea may accompany large or rapidly growing tumors.
Anaplastic presentation: ATC typically presents dramatically as a rapidly enlarging, often rock-hard neck mass that may double in size within days to weeks, accompanied by early onset of hoarseness, dysphagia, and respiratory compromise. This fulminant presentation in an elderly patient with a pre-existing thyroid nodule or known thyroid cancer should prompt immediate evaluation and urgent multidisciplinary management.
MTC-specific features: MTC may cause flushing and diarrhea due to calcitonin and other peptide secretion by the tumor. In the context of MEN2A, concurrent pheochromocytoma (which must be excluded and treated before thyroid surgery to prevent hypertensive crisis) and hyperparathyroidism may be present. MEN2B patients have a characteristic phenotype including marfanoid habitus, mucosal neuromas on the tongue and lips, and intestinal ganglioneuromatosis causing bowel dysmotility.
Systemic symptoms from metastases: Bone pain or pathologic fracture from skeletal metastases (particularly FTC), cough or dyspnea from pulmonary metastases, or neurological symptoms from brain metastases may represent the initial presentation of advanced differentiated or medullary thyroid cancer, though this is uncommon.
Diagnosis and Staging
The evaluation of a thyroid nodule follows a structured algorithmic approach that integrates clinical risk assessment, ultrasound characterization, cytopathology, and increasingly, molecular testing.
Ultrasound Risk Stratification
Thyroid ultrasound is the cornerstone of nodule evaluation, providing information on echogenicity, composition (solid vs. cystic), shape, margins, internal calcifications, vascularity, and size. Several standardized risk stratification systems have been developed to guide the decision to perform fine-needle aspiration biopsy.
The ACR TI-RADS (Thyroid Imaging, Reporting and Data System) assigns a numeric score (1–5) based on five ultrasound categories, translating into FNA recommendations based on both category and nodule size. The ATA (American Thyroid Association) system uses five risk categories (very low, low, intermediate, high, and not applicable/purely cystic), also linking to size-based FNA thresholds. High-suspicion ultrasound features include marked hypoechogenicity, irregular margins, microcalcifications, taller-than-wide shape (on transverse view), and extrathyroidal extension. The point-of-care value of these systems is harmonizing clinical decision-making and reducing unnecessary biopsies of benign nodules while reliably identifying those requiring sampling.
Fine-Needle Aspiration Cytology and the Bethesda System
Ultrasound-guided fine-needle aspiration (FNA) cytology is the primary diagnostic tool for evaluating thyroid nodules meeting biopsy criteria. The 2017 Bethesda System for Reporting Thyroid Cytopathology provides a six-category classification that communicates both diagnosis and malignancy risk:
- Bethesda I (Nondiagnostic/Unsatisfactory): Inadequate sample; repeat FNA recommended. Malignancy risk 1–4%.
- Bethesda II (Benign): Includes benign follicular nodule, colloid nodule, Hashimoto’s thyroiditis, and cystic lesions. Malignancy risk 0–3%; clinical and ultrasound surveillance appropriate.
- Bethesda III (Atypia of Undetermined Significance / Follicular Lesion of Undetermined Significance, AUS/FLUS): Malignancy risk 10–30%; molecular testing strongly recommended to guide management.
- Bethesda IV (Follicular Neoplasm / Suspicious for Follicular Neoplasm): Malignancy risk 25–40%; molecular testing or diagnostic lobectomy depending on clinical context.
- Bethesda V (Suspicious for Malignancy): Malignancy risk 50–75%; surgery recommended.
- Bethesda VI (Malignant): Malignancy risk 97–99%; surgery required.
Molecular Testing for Indeterminate Nodules
For cytologically indeterminate nodules (Bethesda III and IV), molecular testing of FNA material can substantially refine malignancy risk and guide surgical decision-making, potentially avoiding diagnostic lobectomy in patients with benign or very low-risk molecular results. Two commercially available platforms dominate clinical practice:
ThyroSeq v3 (CBLPath/University of Pittsburgh) is a next-generation sequencing-based panel detecting mutations and gene fusions in more than 112 cancer-relevant genes. It has a negative predictive value (NPV) exceeding 96 percent for Bethesda III/IV nodules, making a negative result reassuring enough to support active surveillance over surgery in many cases. The Afirma Gene Expression Classifier (GEC; Veracyte) uses a machine-learning algorithm applied to a 167-gene expression profile, classifying nodules as “benign” (NPV ~94%) or “suspicious.” A newer Afirma Genomic Sequencing Classifier (GSC) combines expression analysis with mutation detection and has improved specificity.
Staging and Advanced Imaging
For clinically apparent or confirmed thyroid cancer, staging workup varies by subtype and presumed extent. Neck ultrasound including central and lateral compartments evaluates lymph node involvement. For DTC suspected to have extrathyroidal extension, involvement of central compartment nodes, or distant metastatic disease, CT of the neck and chest (with iodinated contrast used judiciously — iodine loading delays subsequent RAI therapy) or MRI of the neck provides more detailed anatomic information. PET-CT with FDG is most useful for RAI-refractory DTC and for MTC/ATC, where FDG-avid disease reflects loss of differentiation. Diagnostic whole-body RAI scan with low-dose I-131 or I-123 can identify functioning metastases in DTC after thyroidectomy.
AJCC 8th edition staging for DTC separates patients by age at diagnosis: all patients under 55 are Stage I (no distant metastases) or Stage II (with distant metastases), reflecting the excellent prognosis in young patients regardless of nodal involvement or local extension. Patients 55 and older are staged I through IVB based on tumor size, extrathyroidal extension, and nodal/distant metastases. For MTC and ATC, separate staging algorithms apply, with ATC classified Stage IV regardless of disease extent.
All newly diagnosed MTC patients should undergo germline RET mutation testing. Prior to MTC surgery, plasma or urine catecholamines and metanephrines must be measured to exclude coexistent pheochromocytoma, which requires adrenalectomy before thyroidectomy to avoid intraoperative hypertensive crisis. Serum calcium and PTH evaluate for concurrent hyperparathyroidism in MEN2A.
Treatment
Treatment of thyroid cancer is highly subtype- and stage-dependent, requiring multidisciplinary input from endocrinology, surgery, nuclear medicine, radiation oncology, and medical oncology.
Surgery
Surgery is the primary treatment for all resectable thyroid cancers. For differentiated thyroid cancer, two main surgical options exist: total thyroidectomy (removal of the entire thyroid gland) and thyroid lobectomy (removal of one lobe and the isthmus). Contemporary ATA guidelines support lobectomy as adequate surgical treatment for low-risk papillary thyroid cancer ≤4 cm confined to the thyroid without high-risk features, provided the patient does not require RAI therapy for staging or treatment. Total thyroidectomy is preferred for tumors larger than 4 cm, bilateral disease, significant extrathyroidal extension, known distant metastases, or when RAI ablation is planned.
Central neck dissection (removal of level VI lymph nodes in the central compartment) is recommended for clinically involved central nodes. Prophylactic central dissection in clinically node-negative patients remains controversial, as it may improve staging accuracy but is associated with increased risk of hypoparathyroidism and RLN injury without a proven survival benefit in low-risk cases. Lateral neck dissection (levels II–V) is performed for biopsy-confirmed lateral cervical node involvement.
For ATC, emergent surgery may be necessary for airway compromise (tracheostomy), and resection of resectable Stage IVA disease is pursued when feasible, but the majority of patients present with unresectable locally advanced or metastatic disease. For MTC, total thyroidectomy with central compartment dissection is standard, with lateral dissection guided by preoperative calcitonin levels and imaging.
Radioiodine (RAI) Therapy
Radioiodine (I-131) therapy exploits NIS-mediated iodine uptake in DTC cells to deliver cytotoxic beta-radiation. Following total thyroidectomy, RAI is used for remnant ablation (eliminating residual normal thyroid tissue to enable accurate Tg monitoring), adjuvant treatment in intermediate/high-risk patients, or therapy for known metastatic disease. RAI effectiveness requires adequate TSH stimulation (TSH >30 mIU/L) achieved either by thyroid hormone withdrawal (4–6 weeks of hypothyroidism) or by recombinant human TSH (rhTSH, Thyrogen) injection, which is better tolerated.
Low-risk DTC patients who undergo total thyroidectomy with complete resection of small tumors do not require RAI. Intermediate-risk patients with microscopic extrathyroidal extension, lymph node involvement, or vascular invasion receive RAI to eliminate potential residual disease. High-risk patients with gross extrathyroidal extension, incomplete resection, or distant metastases receive therapeutic high-dose RAI as part of initial treatment.
RAI-refractory DTC — defined by lack of RAI uptake in metastatic lesions, progression on RAI, or cumulative RAI dose limiting further therapy — requires systemic treatment. The transition to RAI-refractory status is associated with tumor dedifferentiation and loss of NIS expression, often linked to BRAF V600E or RAS mutations.
TSH Suppression Therapy
Following thyroidectomy for DTC, levothyroxine therapy serves dual purposes: replacing thyroid hormone function and, when dosed to supraphysiologic levels, suppressing pituitary TSH secretion below the normal range. TSH-independent signaling has been shown to stimulate DTC cell growth in laboratory studies, and retrospective data support TSH suppression in high-risk patients. The degree of suppression is tailored to individual risk: high-risk patients target TSH <0.1 mIU/L initially, while low-risk patients in remission may be maintained at low-normal TSH (0.5–2 mIU/L) to minimize long-term risks of atrial fibrillation, osteoporosis, and cardiac effects from subclinical thyrotoxicosis.
Systemic Therapy for Advanced and RAI-Refractory DTC
The approval of multikinase inhibitors transformed the treatment landscape for progressive RAI-refractory DTC. Lenvatinib (Eisai/Merck) demonstrated a median progression-free survival of 18.3 months versus 3.6 months for placebo in the pivotal SELECT phase III trial (PMID 25671254), with an objective response rate of 65 percent. Sorafenib (Bayer) showed a 10.8-month improvement in PFS versus placebo in the DECISION trial (PMID 24768112) with response rates of 12 percent but disease stabilization in the majority. Lenvatinib is now preferred as first-line therapy for RAI-refractory DTC given its superior efficacy, with sorafenib as an alternative.
For BRAF V600E-mutant RAI-refractory DTC, the combination of BRAF inhibitor dabrafenib plus MEK inhibitor trametinib has shown activity. Additionally, “redifferentiation” strategies using selumetinib (MEK inhibitor) or dabrafenib plus trametinib before RAI can restore radioiodine uptake in a proportion of RAI-refractory tumors, allowing effective RAI retreatment — an approach now supported by phase 2 data.
RET-Targeted Therapy for MTC
The identification of RET as the dominant driver of both hereditary and sporadic MTC, and as a recurrent oncogene in PTC (via RET/PTC fusions), spurred the development of highly selective, potent RET kinase inhibitors. Selpercatinib (Loxo Oncology/Eli Lilly) achieved overall response rates of 69–73 percent in RET-mutant MTC in the LIBRETTO-001 trial (PMID 32846060), with a complete response rate of 10 percent and responses in patients previously treated with vandetanib or cabozantinib. Pralsetinib (Blueprint Medicines) similarly demonstrated response rates of 60–71 percent in RET-mutant MTC. Both agents are now FDA-approved and recommended as preferred first-line therapy for advanced/metastatic RET-mutant MTC, replacing the earlier multikinase inhibitors vandetanib and cabozantinib for this population.
Vandetanib (AstraZeneca) and cabozantinib (Exelixis), which inhibit RET along with VEGFR and other kinases, remain options for RET wild-type MTC or following progression on selective RET inhibitors. Cytotoxic chemotherapy has limited activity in MTC.
Treatment of Anaplastic Thyroid Cancer
ATC treatment requires emergency multidisciplinary coordination. BRAF V600E mutation status should be determined within 24–72 hours of diagnosis when possible, as it identifies patients eligible for the most effective treatment. The combination of dabrafenib (BRAF inhibitor) plus trametinib (MEK inhibitor) produces an overall response rate of approximately 69 percent in BRAF V600E-mutant ATC and is associated with long-term remissions in a small subset of patients — a remarkable result in a disease previously uniformly fatal within weeks (PMID 30267869). This regimen is now recommended as initial systemic therapy for all BRAF V600E-mutant ATC, concurrent with consideration of surgery and/or radiotherapy for resectable Stage IVA/B disease.
For BRAF wild-type ATC, pembrolizumab (PD-1 checkpoint inhibitor) has demonstrated activity with an objective response rate of approximately 19 percent in small series, and combination immunotherapy and chemotherapy regimens are under investigation. External beam radiotherapy (EBRT) to the neck is an important component of ATC management for local disease control, often given concurrently with radiosensitizing systemic therapy. Despite these advances, ATC remains largely incurable in most patients, and goals-of-care discussions are an essential early part of management.
Prognosis
Prognosis in thyroid cancer varies enormously by subtype and stage, with differentiated thyroid cancers carrying some of the most favorable survival outcomes among all malignancies and anaplastic cancer representing the opposite extreme.
Differentiated Thyroid Cancer
The 10-year disease-specific survival for DTC overall exceeds 95 percent, with excellent outcomes even for patients with lymph node metastases. The risk of disease recurrence, however, is more nuanced and is the primary focus of risk stratification tools. The ATA risk stratification system (low, intermediate, high) applied after initial surgery and pathology review guides the intensity of follow-up, TSH suppression targets, and RAI use.
Low-risk patients — with small, intrathyroidal PTC without aggressive histology, no vascular invasion, no lymph node involvement — have recurrence rates under 5 percent at 10 years. Intermediate-risk features (minor extrathyroidal extension, modest nodal involvement, vascular invasion) correspond to 5–20 percent recurrence rates. High-risk patients (gross extrathyroidal extension, incomplete resection, extensive nodal involvement with extranodal extension, or distant metastases) have substantially higher recurrence rates.
The MACIS scoring system (Metastasis, Age, Completeness of resection, Invasion, Size) provides an older but validated numeric estimate of DTC mortality risk from the Mayo Clinic experience and remains useful in clinical practice. Dynamic risk stratification — reassessing response to initial therapy at 12–18 months using serum Tg, TgAb trends, and neck ultrasound — allows reclassification of initial risk estimates and further individualization of follow-up intensity.
Age is a powerful prognostic factor unique to thyroid cancer: the AJCC 8th edition system classifies all patients under 55 as Stage I or II regardless of tumor size or nodal involvement, reflecting the consistently excellent outcomes in younger patients. Beyond age 55, stage increases with extent of disease, and outcomes worsen progressively.
Medullary Thyroid Cancer
MTC prognosis is strongly stage-dependent. Patients with disease confined to the thyroid (Stage I/II) have 10-year survival rates of approximately 75–95 percent. Locally advanced disease with nodal involvement has intermediate outcomes. Distant metastatic MTC has a 10-year survival of approximately 20 percent, though outcomes are improving with effective RET-targeted therapies. Calcitonin and CEA doubling times are the strongest biochemical predictors of survival — calcitonin doubling time under 6 months carries a 5-year survival of approximately 25 percent, while doubling time over 24 months correlates with near-normal 10-year survival.
Patients with hereditary MTC identified through genetic screening who undergo prophylactic thyroidectomy before calcitonin becomes detectable have excellent long-term outcomes with surgical cure in the majority. This represents a successful application of genetic risk stratification to cancer prevention.
Anaplastic Thyroid Cancer
ATC median overall survival is 3–5 months from diagnosis in historical series. Even with aggressive multimodality treatment, fewer than 10 percent of patients survive 2 years. However, the BRAF V600E subgroup treated with dabrafenib plus trametinib represents a meaningful exception — durable responses exceeding 2 years have been reported in individual patients, and the 1-year overall survival in BRAF-mutant patients treated with this combination is approximately 80 percent in some series, compared to historical rates under 20 percent. These outcomes, while not curative for most, represent a paradigm shift in ATC management and support molecular profiling at diagnosis in every patient.
Factors associated with modestly better ATC outcomes include disease confined to the thyroid (Stage IVA), younger age, and BRAF V600E mutation (due to targeted therapy availability). The presence of a coexisting differentiated component may paradoxically indicate slightly better prognosis, as these tumors may retain some responsiveness to RAI or have a less completely dedifferentiated biology.
Key Research Papers
- Haugen BR, Alexander EK, Bible KC, et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016;26(1):1–133. PMID 26462967
- Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer (SELECT Trial). N Engl J Med. 2015;372(7):621–630. PMID 25671254
- Brose MS, Nutting CM, Jarzab B, et al. Sorafenib in Radioactive Iodine-Refractory, Locally Advanced or Metastatic Differentiated Thyroid Cancer (DECISION): a randomised, double-blind, phase 3 trial. Lancet. 2014;384(9940):319–328. PMID 24768112
- Subbiah V, Wirth LJ, Smit EF, et al. Selpercatinib in Patients with RET-Mutant Medullary Thyroid Cancer (LIBRETTO-001). N Engl J Med. 2020;383(9):825–835. PMID 32846060
- Shah MH, Lombard-Bohas C, Samuels E, et al. Selpercatinib in Patients with Previously Treated RET-Mutant Medullary Thyroid Cancer. J Clin Oncol. 2021;39(34):3851–3861. — Search PubMed
- Subbiah V, Cabanillas ME, Cote GJ, et al. Dabrafenib Plus Trametinib in BRAF V600E-Mutated Anaplastic Thyroid Cancer. N Engl J Med. 2018;379(13):1377. — Search PubMed
- Fagin JA, Wells SA Jr. Biologic and Clinical Perspectives on Thyroid Cancer. N Engl J Med. 2016;375(23):2307–2316. — Search PubMed
- Sipos JA, Mazzaferri EL. Thyroid Cancer Epidemiology and Prognostic Variables. Clin Oncol (R Coll Radiol). 2010;22(6):395–404. — Search PubMed
- Tessler FN, Middleton WD, Grant EG, et al. ACR Thyroid Imaging, Reporting and Data System (TI-RADS): White Paper of the ACR TI-RADS Committee. J Am Coll Radiol. 2017;14(5):587–595. PMID 28372962
- Cibas ES, Ali SZ. The 2017 Bethesda System for Reporting Thyroid Cytopathology. Thyroid. 2017;27(11):1341–1346. PMID 29091573
- Wells SA Jr, Asa SL, Dralle H, et al. Revised American Thyroid Association Guidelines for the Management of Medullary Thyroid Carcinoma. Thyroid. 2015;25(6):567–610. PMID 25810047
- Smallridge RC, Ain KB, Asa SL, et al. American Thyroid Association Guidelines for Management of Patients with Anaplastic Thyroid Cancer. Thyroid. 2012;22(11):1104–1139. PMID 23130564
Connections
- Cancer (Overview)
- Oncology
- Head and Neck Cancer
- Endocrinology
- Parathyroid Disorders
- Hyperthyroidism
- Hypothyroidism
- Multiple Endocrine Neoplasia (MEN2)
- Iodine
- Lab Tests (TSH, Thyroglobulin)