Pheochromocytoma
A pheochromocytoma is a tumor that grows inside the adrenal gland — the small hat-shaped organ that sits on top of each kidney — and releases massive, uncontrolled bursts of adrenaline-like hormones called catecholamines. Imagine your body's fight-or-flight response firing at full blast without any actual danger: blood pressure can spike to stroke-level heights in minutes, your heart pounds, your skin drips with sweat, and a crushing headache arrives out of nowhere. For the roughly 1 in 500,000 people diagnosed each year, these episodes are terrifying and baffling until the diagnosis is finally made. What makes the condition genuinely dangerous is that it can stay hidden for years — only to declare itself fatally during surgery, childbirth, or a routine anesthetic induction if the surgical team is not prepared. The good news is that once the diagnosis is secured through blood and urine tests, careful pre-operative medication can blunt the hormone surges completely, and laparoscopic surgery is then curative in the vast majority of cases. The key is knowing to look for it.
Table of Contents
- What a Pheochromocytoma Is
- The "Rule of 10s" — Updated for the Genomic Era
- Classic Symptoms and Paroxysmal Crises
- Hereditary Syndromes and Germline Mutations
- Biochemical Diagnosis
- Imaging and Localization
- Pre-operative Management — Why Alpha-Blockade First
- Surgical Treatment
- Malignant Pheochromocytoma and Paraganglioma
- Research Papers
- Connections
- Featured Videos
What a Pheochromocytoma Is
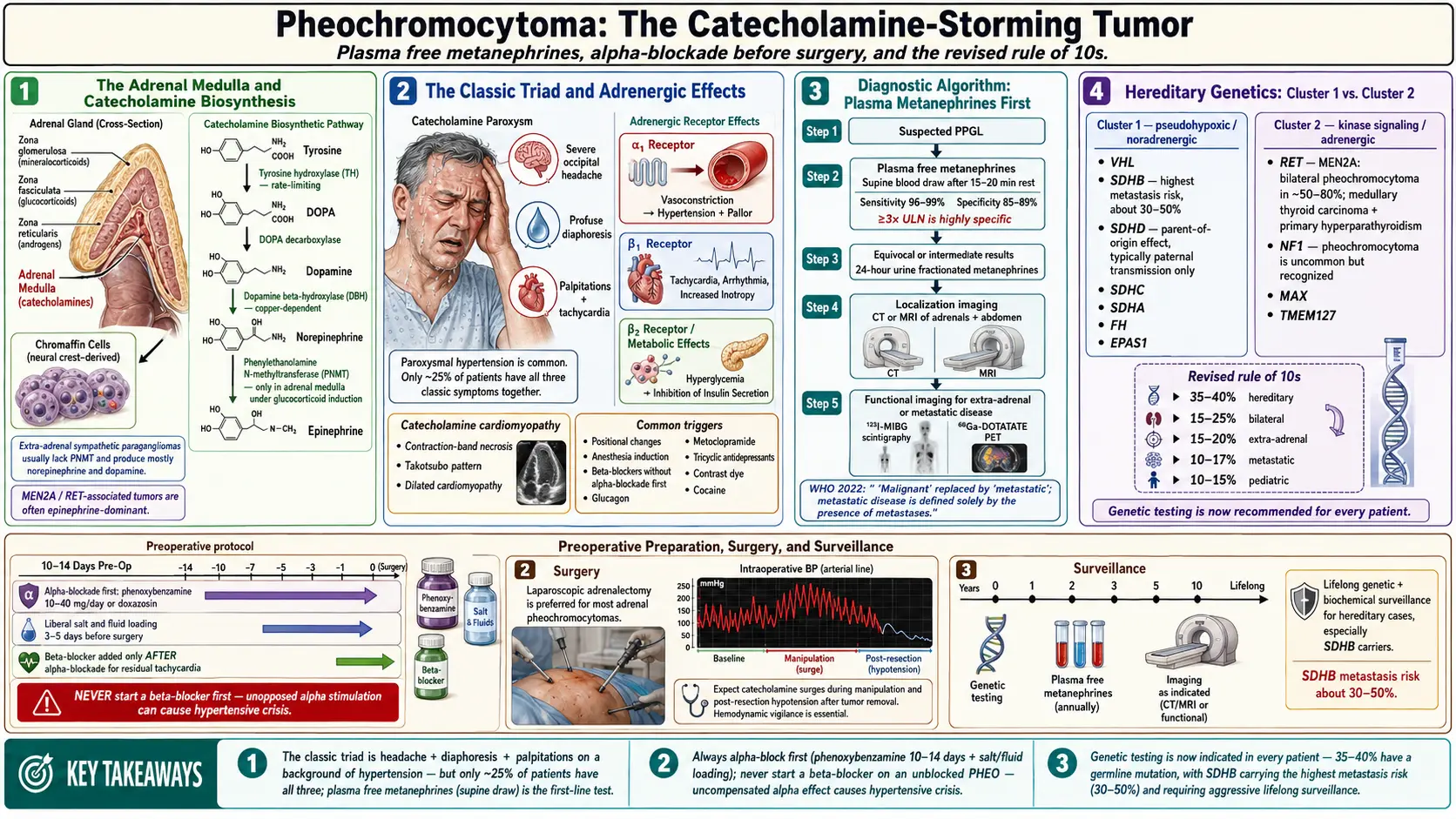
Your adrenal medulla — the inner core of each adrenal gland — is built from a specialized type of cell called a chromaffin cell. These cells are the body's fast-response adrenaline factory: under normal circumstances they release precisely calibrated pulses of epinephrine (adrenaline) and norepinephrine when you need them. A pheochromocytoma is a tumor that arises from these chromaffin cells and loses the ability to regulate its own output. It can release enormous quantities of catecholamines spontaneously or in response to triggers, producing a cardiovascular stress response that the body was never designed to sustain.
Approximately 80% of pheochromocytomas arise in the adrenal medulla itself. The remaining 20% grow from chromaffin tissue outside the adrenal glands; these are called paragangliomas. The most common extra-adrenal site is the organ of Zuckerkandl, a small cluster of chromaffin tissue near the aortic bifurcation at the level of the inferior mesenteric artery. Other sites include the carotid body (carotid paraganglioma), the jugulotympanic region of the skull base, the bladder wall (which causes catecholamine release precisely during urination — a diagnostic clue), the mediastinum, and scattered retroperitoneal sites along the sympathetic chain.
The term pheochromocytoma literally means "dusky-colored tumor" — a reference to the dark staining these tumors produce when exposed to chromium salts, which was how they were first recognized histologically in the early 20th century. Today the diagnosis is biochemical and radiological, but the name has stuck.
Most pheochromocytomas are benign in the sense that they do not spread to distant organs. However, even a benign tumor causes major harm through the cardiovascular effects of the catecholamines it secretes. Undetected, long-standing pheo can cause hypertensive cardiomyopathy (a heart that has been overworked for years), hemorrhagic stroke, and sudden death. The danger during surgical procedures under general anesthesia — where airway manipulation and the release of anesthetic agents can provoke a massive catecholamine surge — has been recognized since the first successful surgical removal in 1926.
The "Rule of 10s" — Updated for the Genomic Era
Medical students have been taught for decades to remember pheochromocytoma by the "Rule of 10s": 10% are malignant, 10% are bilateral, 10% are extra-adrenal, 10% occur in children, and 10% are familial (hereditary). This teaching rule was a useful mnemonic for a generation, but genomic medicine has shown it to be a significant underestimate on the hereditary side — and therefore on the malignancy side as well.
The updated picture:
- Familial/hereditary: Originally cited as 10%; now known to be approximately 35–40% of all pheochromocytomas and paragangliomas. Germline testing is now recommended for all patients, regardless of age, family history, or apparent sporadic presentation.
- Malignant (metastatic): Approximately 10–15% for adrenal pheochromocytoma overall, but rises dramatically to 30–50% for paraganglioma with an SDHB mutation. The term "malignant" in this context means metastasis to sites where chromaffin tissue does not normally exist (liver, lung, bone, lymph nodes) — you cannot call a pheo malignant based on histology alone.
- Bilateral: Roughly 10% remain bilateral (both adrenal glands); this proportion is higher in hereditary syndromes such as MEN2 and VHL.
- Extra-adrenal: Roughly 15–20% are extra-adrenal paragangliomas, slightly higher than the traditional 10%.
- Pediatric: About 10–20% of cases are diagnosed in children and adolescents; in this age group, extra-adrenal location and hereditary mutations are even more common than in adults.
The practical lesson from updating the Rule of 10s: every patient with a pheochromocytoma or paraganglioma should be offered germline genetic testing, because the mutation found determines malignancy risk, the need for surveillance of other organs, and whether first-degree relatives should be tested.
Classic Symptoms and Paroxysmal Crises
The cardinal clinical signature of pheochromocytoma is the paroxysmal hypertensive crisis: a sudden, dramatic spike in blood pressure — sometimes exceeding 250/150 mmHg — that arrives without warning, lasts minutes to an hour, and then fades. Between episodes, blood pressure may be normal or only mildly elevated, which is why routine measurements often miss the diagnosis entirely.
The classic symptom triad, present in roughly two thirds of patients with a paroxysm, is:
- Severe headache — present in approximately 80% of symptomatic patients; typically pounding/throbbing, reflecting the rapid blood pressure rise
- Diaphoresis (profuse sweating) — present in approximately 71%; catecholamines activate sweat glands directly
- Palpitations — present in approximately 64%; tachycardia is driven by both epinephrine (increases heart rate) and the reflex response to hypertension
When this triad occurs together with paroxysmal or sustained hypertension, pheochromocytoma should be high on the differential. Other associated symptoms include pallor (vasoconstriction), anxiety or a sense of impending doom, tremor, nausea, and chest or abdominal pain. Some patients describe a "spell" lasting 15–20 minutes during which they feel as if they might die — and then feel completely normal afterward. These spells are often dismissed as panic attacks for years before the correct diagnosis is made.
Triggers that provoke a paroxysm include:
- Physical pressure on the tumor — bending, straining, or direct palpation of the abdomen; this is why a doctor performing an abdominal exam can inadvertently trigger a crisis
- Anesthetic induction and airway manipulation — laryngoscopy and intubation are classic triggers; endotracheal intubation without prior alpha-blockade can be fatal
- Tyramine-rich foods — aged cheeses, red wine, cured meats; normally catabolized by MAO in the gut and liver, but the high catecholamine environment in pheo can overwhelm this system
- Beta-blockers given first — this is a critical clinical trap. If a beta-blocker is prescribed for the hypertension before adequate alpha-blockade is in place, it blocks the vasodilatory beta-2 receptors in peripheral vessels while leaving alpha-1 (vasoconstrictor) receptors unopposed. The result is an extreme, sometimes fatal hypertensive crisis
- Glucagon — used in radiology procedures or as a GI antispasmodic, it directly stimulates catecholamine release from the tumor
- Micturition (urination) — a bladder paraganglioma releases catecholamines when the bladder contracts; this is essentially pathognomonic for bladder pheo
- Childbirth — uterine contractions compress abdominal tissue; undiagnosed pheo is a leading cause of maternal death in pregnancy-associated hypertensive emergencies
- Metoclopramide and other dopamine-blocking drugs can provoke catecholamine release
Approximately 10–15% of patients are asymptomatic — their tumor is discovered incidentally during imaging performed for another reason (an "adrenal incidentaloma"). This is increasingly common as CT and MRI use has grown.
Hereditary Syndromes and Germline Mutations
Pheochromocytoma and paraganglioma are among the most heritable of all human tumors. At least 16 susceptibility genes have now been identified, and approximately 35–40% of patients carry a germline (inherited) pathogenic variant. The major syndromes and genes are:
SDH genes (succinate dehydrogenase complex subunits): SDHB, SDHC, SDHD, and SDHAF2 encode subunits of mitochondrial complex II. Loss of function causes accumulation of succinate, which inhibits prolyl hydroxylases and stabilizes HIF-1alpha — the hypoxia-response transcription factor — creating a "pseudohypoxia" state that drives tumor formation. SDHB mutations carry the highest malignancy risk (30–50% for paraganglioma), and every SDHB-mutant patient warrants surveillance imaging every 1–2 years for life. SDHD mutations tend to cause head and neck paragangliomas and show genomic imprinting (only maternal-copy mutations are pathogenic; paternal copies are silenced).
RET proto-oncogene — Multiple Endocrine Neoplasia type 2 (MEN2):
- MEN2A: pheochromocytoma (50% lifetime risk) + medullary thyroid carcinoma (near 100% penetrance, usually the presenting tumor) + primary hyperparathyroidism. RET codon 634 is the most common mutation.
- MEN2B: pheochromocytoma + medullary thyroid carcinoma (most aggressive form, often presenting in infancy) + mucosal neuromas of the tongue and lips + Marfanoid habitus. RET codon 918 (M918T) accounts for 95% of MEN2B. These patients often go undiagnosed for years because the mucosal neuromas are subtle.
In MEN2, pheochromocytomas are almost always epinephrine-secreting (unlike the norepinephrine predominance seen in SDH mutations) and are usually bilateral but rarely malignant.
VHL gene — Von Hippel-Lindau disease: VHL encodes a protein that targets HIF-1alpha for ubiquitin-mediated degradation. Loss of VHL function stabilizes HIF-1alpha, driving tumor formation throughout highly vascularized tissues. Clinical features include pheochromocytoma + hemangioblastomas of the cerebellum, brainstem, and spinal cord + clear cell renal cell carcinoma + pancreatic neuroendocrine tumors + endolymphatic sac tumors. VHL-associated pheos tend to be norepinephrine-secreting paragangliomas.
NF1 — Neurofibromatosis type 1: NF1 encodes neurofibromin, a RAS-GTPase-activating protein. Clinical hallmarks include cafe-au-lait macules (≥6, >1.5 cm after puberty), axillary/inguinal freckling, Lisch nodules on the iris, and neurofibromas. Pheochromocytoma occurs in roughly 0.1–5.7% of NF1 patients but is systematically under-screened. NF1-associated pheos are almost always adrenal, unilateral, and epinephrine-secreting.
MAX and TMEM127 are more recently identified susceptibility genes associated with bilateral adrenal pheo in younger patients, with moderate malignancy risk. TMEM127 mutations are particularly associated with adrenal tumors with a norepinephrine phenotype.
For clinical practice: the Endocrine Society guideline recommends that all patients with pheochromocytoma or paraganglioma be offered germline testing by a genetic counselor, with panel testing covering at minimum SDHB/C/D/AF2, RET, VHL, NF1, MAX, and TMEM127. A positive finding changes the surveillance plan for the patient and their family.
Biochemical Diagnosis
The biochemical cornerstone of the diagnosis is measuring metanephrines — the metabolic breakdown products of epinephrine (metanephrine) and norepinephrine (normetanephrine). Pheochromocytoma continuously metabolizes its own catecholamines even between symptomatic crises, producing a persistently elevated baseline of metanephrines that is far more reliable than measuring the catecholamines themselves, which spike and fall rapidly.
Plasma free metanephrines (measured by LC-MS/MS or immunoassay from a fasting blood draw) are the test of choice for screening. In the 2014 Endocrine Society guideline, sensitivity for pheochromocytoma/paraganglioma is approximately 97% with a specificity of approximately 85%. This high sensitivity means a normal result essentially rules out the tumor in most clinical settings — but the 15% false-positive rate means that mildly elevated results must be interpreted carefully before proceeding to imaging.
24-hour urine metanephrines and catecholamines provide slightly lower sensitivity (~87%) but higher specificity (~99%), making them useful for confirmatory testing when plasma metanephrines are borderline elevated. The patient collects all urine over 24 hours into an acidified container; the sample is then measured for fractionated metanephrines and creatinine (to verify adequate collection).
Important sources of false-positive elevations that must be excluded before pursuing imaging:
- Tricyclic antidepressants (amitriptyline, nortriptyline) — block norepinephrine reuptake, dramatically elevating normetanephrine
- Serotonin-norepinephrine reuptake inhibitors (SNRIs) such as venlafaxine and duloxetine — similar mechanism
- Sympathomimetics — decongestants, amphetamines, cocaine
- Levodopa — metabolized to dopamine and then catecholamines
- Acetaminophen (paracetamol) — interferes with some immunoassay platforms, falsely elevating plasma metanephrine on those particular assays (this is assay-specific; LC-MS/MS platforms are not affected)
- Physiologic stress — severe illness, surgery, obstructive sleep apnea, and intense exercise can all raise catecholamine output modestly
When plasma free metanephrines are markedly elevated (more than four times the upper limit of normal), the specificity for pheochromocytoma is essentially 100% and the diagnosis can be considered confirmed biochemically pending localization imaging.
Plasma methoxytyramine — the O-methylated metabolite of dopamine — is increasingly measured alongside metanephrines. Elevated methoxytyramine is associated with dopamine-secreting tumors, which in turn are associated with SDHB mutations and higher malignancy risk. It can also help localize head-and-neck paragangliomas that do not secrete norepinephrine or epinephrine.
Imaging and Localization
Imaging is performed only after biochemical confirmation. The sequence chosen depends on tumor size, location, and genetic subtype.
CT of the adrenal glands is the first-line anatomical imaging modality. Pheochromocytomas are typically large (>4 cm, though some are discovered smaller on incidental imaging), heterogeneous (areas of necrosis and hemorrhage are common), and show high-density enhancement after IV contrast. The adrenal washout characteristics used to characterize incidentalomas — an absolute washout >60% at 15 minutes is reassuring for adenoma — should NOT be used to evaluate a suspected pheo: the contrast itself can trigger a catecholamine crisis in an unprepared patient. CT is excellent for adrenal tumors but less sensitive for small extra-adrenal paragangliomas.
MRI has two key advantages over CT: no ionizing radiation (important for young patients and for surveillance), and characteristic T2 signal. Pheochromocytomas appear intensely bright on T2-weighted sequences — the so-called "light bulb sign" — due to their high water content and vascular nature. MRI is preferred for pregnant patients, for extra-adrenal tumors along the sympathetic chain, and for patients in whom repeat imaging will be needed frequently.
Functional (nuclear medicine) imaging is used when:
- The CT/MRI is negative despite high biochemical suspicion (extra-adrenal or small primary)
- Metastatic disease is suspected
- Pre-operative planning for possible MIBG therapy
Available functional modalities:
- 123I-MIBG scintigraphy (meta-iodobenzylguanidine): MIBG is a norepinephrine analogue that is taken up by functioning adrenergic tissue. Traditional workhorse for extra-adrenal and metastatic disease; sensitivity ~75–90% for adrenal pheo, lower for SDH-related paragangliomas which often lack the norepinephrine transporter (NET) needed for uptake
- 68Ga-DOTATATE PET/CT (Gallium-68-labeled somatostatin analogue): now the preferred functional imaging modality for SDH-related paragangliomas, which overexpress somatostatin receptors. Sensitivity exceeds 90% for these tumors and it provides superior spatial resolution to MIBG scintigraphy
- 18F-FDOPA PET (fluorine-18-labelled DOPA): excellent for extra-adrenal paragangliomas with a norepinephrine phenotype; less useful for SDHB-mutant tumors
- 18F-FDG PET/CT: used primarily for staging malignant/metastatic disease; SDHB-mutant paragangliomas are often FDG-avid
The choice of functional scan is therefore guided by the germline mutation: 68Ga-DOTATATE for SDH mutations, MIBG for sporadic and RET/VHL-related pheo, and FDG for suspected metastatic SDHB.
Pre-operative Management — Why Alpha-Blockade First
The most important principle in preparing a patient for pheochromocytoma surgery is alpha-adrenergic blockade first, for a minimum of 10–14 days before the operation. Skipping or abbreviating this step — or reversing the order by adding a beta-blocker first — has killed patients.
Here is what happens if you do not pre-treat: the surgeon's first manipulation of the tumor, or anesthetic induction, releases a massive bolus of catecholamines directly into the bloodstream. Blood pressure can reach 300 mmHg or more; the result can be hypertensive encephalopathy, hemorrhagic stroke, myocardial infarction from coronary vasospasm, or fatal cardiac arrhythmia. The anesthesiologist and surgeon are managing a cardiovascular emergency with no warning.
Alpha-blockade works by occupying alpha-adrenergic receptors in blood vessel walls before the catecholamine surge arrives, so when catecholamines are released intraoperatively there is nothing for them to bind to cause vasoconstriction. Equally important, by relaxing peripheral vessels over the preoperative days, it allows intravascular volume to expand. Patients with long-standing pheo are severely intravascularly volume-depleted because chronic alpha-adrenergic stimulation has kept their vessels clamped; reversal of this vasoconstriction unmasks a relative hypovolemia that must be corrected before surgery or profound hypotension will follow tumor ligation.
Phenoxybenzamine is the traditional agent of choice: it is an irreversible, non-selective alpha-1 and alpha-2 blocker. Starting dose is 10 mg twice daily, titrated every 2–3 days by 10–20 mg increments until blood pressure is well controlled (goal: sitting BP <130/80 mmHg, standing BP >80 mmHg systolic to avoid syncope). Doses of 20–100 mg daily are common. Because it is irreversible and non-selective, it also blocks alpha-2 receptors (which normally suppress norepinephrine release), which means it can cause substantial reflex tachycardia and nasal congestion — these side effects are expected and confirm that the drug is working.
Selective alpha-1 blockers (doxazosin, prazosin, terazosin) are reversible and more selective, causing fewer side effects and less reflex tachycardia. Many endocrinologists prefer them in current practice. However, because they are reversible, intraoperative catecholamine surges may partially displace them, meaning the anesthesiologist must be prepared to escalate IV alpha-blockade rapidly.
Beta-blockers are added only after at least 3–5 days of adequate alpha-blockade, solely to control the reflex tachycardia that alpha-blockade induces. Never start a beta-blocker first: in the unblocked state, catecholamines acting on beta-2 receptors in peripheral vessels cause vasodilation that partially offsets alpha-mediated constriction. Remove that vasodilatory tone with a beta-blocker — and you get unopposed alpha stimulation and a catastrophic hypertensive emergency. This rule is one of the most important pharmacological safety principles in endocrine surgery.
The patient is instructed to eat a high-salt, high-fluid diet during the preoperative period to help restore intravascular volume. Target hemodynamic goals assessed 24–48 hours before surgery: no blood pressure readings above 160/90 on 24-hr monitoring, no orthostatic hypotension below 80 mmHg systolic, heart rate 60–70 bpm at rest, no ST-segment changes.
Surgical Treatment
Surgery is the definitive cure. In skilled hands, with appropriate preoperative preparation, outcomes are excellent and operative mortality has fallen to under 3% at major centers.
Laparoscopic adrenalectomy (minimally invasive) is the preferred approach for adrenal pheochromocytomas smaller than approximately 6 cm. The surgeon places three or four small port incisions in the flank or abdomen, introduces a camera, and dissects the adrenal gland free from surrounding tissue before removing it intact. Hospital stay is typically 1–3 days; recovery is 2–4 weeks. The critical surgical rule is to ligate the adrenal vein early and avoid squeezing the gland before the vascular supply is controlled, to minimize intraoperative catecholamine release.
Open adrenalectomy is used for tumors larger than 6 cm, locally invasive or malignant tumors, and cases where anatomy is complex. For large extra-adrenal paragangliomas along the aorta, open surgery via a midline laparotomy is often necessary.
Bilateral pheochromocytoma presents a special challenge: removing both adrenal glands causes permanent primary adrenal insufficiency, requiring lifelong corticosteroid replacement. Some surgeons therefore perform cortical-sparing adrenalectomy — removing the medullary tumor while preserving a rim of functioning adrenal cortex — particularly in hereditary cases (MEN2, VHL) where bilateral disease is expected and the patient will need multiple surgeries over a lifetime. The trade-off is a small but real risk of recurrence from the preserved cortex.
Intraoperative anesthetic management is a critical team sport. The anesthesiologist should have IV phentolamine (a rapid-onset, reversible alpha-blocker) immediately available for blood pressure spikes during manipulation. IV magnesium sulfate is useful as both an antihypertensive and antiarrhythmic. Nicardipine (IV calcium channel blocker) or sodium nitroprusside are alternatives. After the adrenal vein is ligated and the tumor is removed, catecholamine levels fall precipitously within minutes, and the pre-existing volume depletion — even when partially corrected preoperatively — often causes significant hypotension. The anesthesiologist must be ready with IV fluid boluses and vasopressors (norepinephrine infusion is commonly used).
Postoperatively, patients are monitored in ICU or a high-dependency unit for 24 hours. Blood pressure typically normalizes within days to weeks as the catecholamine excess clears. Hypertension persisting beyond 1–3 months after surgery suggests either a missed second tumor, residual disease, or underlying essential hypertension that was unmasked.
Biochemical cure is confirmed at 2–6 weeks post-surgery with repeat plasma free metanephrines. Thereafter, annual biochemical screening is recommended for 10 years (some guidelines say lifelong), because late recurrence or a second primary can occur — particularly in hereditary disease.
Malignant Pheochromocytoma and Paraganglioma
Malignancy in pheochromocytoma and paraganglioma is defined exclusively by metastatic spread to sites where chromaffin tissue does not normally exist — most commonly bone (spine, pelvis, ribs), liver, lungs, and distant lymph nodes. Histological features such as vascular invasion or mitotic figures can suggest higher risk but cannot diagnose malignancy; only distant spread confirms it. The PASS (Pheochromocytoma of the Adrenal gland Scaled Score) and GAPP (Grading system for Adrenal Pheochromocytoma and Paraganglioma) are scoring systems used to stratify risk, but neither is fully reliable.
The single strongest predictor of malignancy is an SDHB germline mutation. Approximately 30–50% of SDHB-mutant paragangliomas will eventually metastasize, compared with roughly 10–15% for adrenal pheochromocytomas overall. For this reason, SDHB-positive patients are placed on intensive long-term surveillance regardless of whether the primary tumor was "successfully" removed.
Treatment options for metastatic pheochromocytoma/paraganglioma:
- Surgery — resection of isolated metastases can prolong survival and improve symptom control, particularly for liver metastases amenable to hepatectomy or ablation
- 131I-MIBG therapy — for tumors that demonstrate MIBG uptake on diagnostic scan. High-specific-activity iobenguane I-131 (Azedra) was FDA-approved in 2018 specifically for unresectable, MIBG-avid metastatic pheochromocytoma and paraganglioma. In the pivotal trial, approximately 25% of patients achieved a durable reduction in antihypertensive medication — the primary endpoint — with an overall disease-control rate near 90%
- 177Lu-DOTATATE (Lutathera) — for DOTATATE-avid tumors (especially SDH-related); approved in other neuroendocrine tumors, used off-label or in trials for paraganglioma
- Sunitinib — a multi-targeted tyrosine kinase inhibitor with activity against VEGFR; used for progressive metastatic disease, particularly SDHB-mutant tumors driven by the pseudohypoxia-VEGF axis
- CVD chemotherapy (cyclophosphamide, vincristine, dacarbazine) — the oldest systemic regimen; partial responses in approximately 50% but complete responses are rare; used when targeted options are exhausted or not available
- Everolimus (mTOR inhibitor) — some activity, particularly in SDH-related disease; ongoing trials
- Cabozantinib — RET/VEGFR/MET inhibitor with particular relevance for RET-driven disease; phase II data suggest activity
Metastatic pheochromocytoma is not curable with current therapies, but disease can be controlled for years. Median survival after diagnosis of metastatic disease is approximately 5 years, though outcomes vary enormously with mutation subtype and extent of disease. All patients with metastatic or high-risk disease should be managed at a center with multidisciplinary expertise and offered enrollment in clinical trials where eligible.
Research Papers
- Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99(6):1915–1942. PMID 24893135. doi:10.1210/jc.2014-2367
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665–675. PMID 16112304. doi:10.1016/S0140-6736(05)67139-5
- Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346(19):1459–1466. PMID 12000816. doi:10.1056/NEJMoa020152
- Fishbein L, Leshchiner I, Walter V, et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell. 2017;31(2):181–193. PMID 28162975. doi:10.1016/j.ccell.2017.01.001
- Eisenhofer G, Lenders JW, Timmers H, et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem. 2011;57(3):411–420. PMID 21097911. doi:10.1373/clinchem.2010.153320
- Pacak K, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. Nat Clin Pract Endocrinol Metab. 2007;3(2):92–102. PMID 17237836. doi:10.1038/ncpendmet0396
- Goldstein RE, O'Neill JA Jr, Holcomb GW 3rd, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755–764. PMID 10366364. doi:10.1097/00000658-199906000-00001
- Pham TH, Moir C, Thompson GB, et al. Pheochromocytoma and paraganglioma in children: a review of medical and surgical management at a tertiary care center. Pediatrics. 2006;118(3):1109–1117. PMID 16951005. doi:10.1542/peds.2005-2299
- Jimenez C, Rohren E, Habra MA, et al. Current and future treatments for malignant pheochromocytoma and sympathetic paraganglioma. Curr Oncol Rep. 2013;15(4):356–371. PMID 23605190. doi:10.1007/s11912-013-0320-x
- Turkova H, Prodanov T, Maly M, et al. Characteristics and Outcomes of Metastatic SDHB and Sporadic Pheochromocytoma/Paraganglioma: A National Institutes of Health Study. Endocr Pract. 2016;22(3):302–314. PMID 26562484. doi:10.4158/EP15725.OR
- Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822–3828. PMID 17652225. doi:10.1210/jc.2007-0709
- Hescot S, Leboulleux S, Amar L, et al. One-year assessment of morphologic and functional changes in metastatic pheochromocytoma and paraganglioma using 131I-metaiodobenzylguanidine therapy. J Clin Endocrinol Metab. 2013;98(11):4464–4471. PMID 23928662. doi:10.1210/jc.2012-3093
Connections
- Paraganglioma — the extra-adrenal chromaffin-cell tumour.
- Adrenal Insufficiency
- Cushing's Syndrome
- Primary Hyperaldosteronism
- Hypertension
- Endocrinology Conditions
- Addison's Disease
- Cortisol Lab Test
- Lab Tests Overview