Diabetes Insipidus
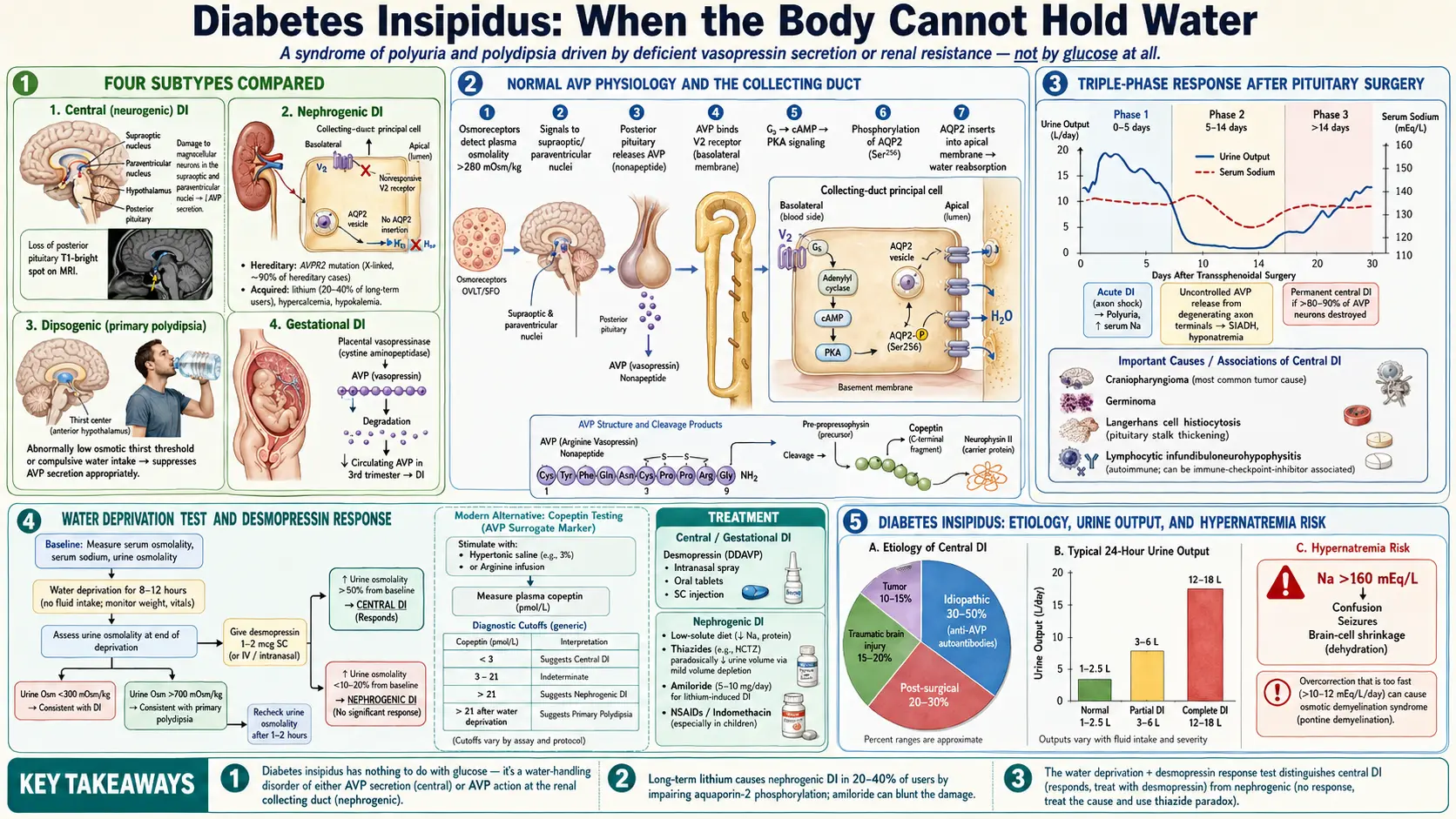
Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research
- References
- Featured Videos
1. Overview
Diabetes insipidus (DI) is a syndrome of impaired water homeostasis characterized by the excretion of large volumes of dilute urine (polyuria) and compensatory polydipsia, resulting from either deficient secretion of arginine vasopressin (AVP; antidiuretic hormone, ADH) or resistance of the renal tubules to its action. The name derives from the Latin insipidus (tasteless), distinguishing the copious, watery (tasteless) urine from the sweet urine of diabetes mellitus — a distinction recognized by clinicians since antiquity and confirmed chemically in the 18th century.
Diabetes insipidus is classified into four principal subtypes based on pathophysiology:
- Central (neurogenic) DI: Deficient AVP synthesis or secretion from the hypothalamic-neurohypophyseal system. Ranges from partial to complete deficiency.
- Nephrogenic DI: Normal or elevated AVP secretion, but the collecting duct principal cells fail to respond appropriately — either due to receptor dysfunction or downstream signaling failure.
- Dipsogenic (primary polydipsia): Excessive water intake suppresses AVP secretion, producing dilute polyuria. AVP secretion is intact; the defect lies in an abnormally low osmotic threshold for thirst or compulsive water drinking.
- Gestational DI: Accelerated AVP degradation by placental vasopressinase (cystine aminopeptidase) during pregnancy, typically presenting in the third trimester.
Accurate subtype classification is essential because treatment differs fundamentally: desmopressin (synthetic AVP analog) is the cornerstone of central and gestational DI management but is ineffective and potentially hazardous in primary polydipsia, and is variably effective in nephrogenic DI. The water deprivation test combined with exogenous desmopressin response remains the traditional diagnostic cornerstone, now increasingly supplemented by the copeptin (C-terminal AVP precursor) stimulation protocol.
2. Epidemiology
- Prevalence: DI is estimated to affect approximately 1 in 25,000 individuals in the general population, though this figure is difficult to validate given underdiagnosis and the heterogeneous etiologic spectrum. In specialized pituitary centers, central DI accounts for 10–15% of patients evaluated for pituitary-hypothalamic disorders.
- Central DI incidence: The overall incidence of central DI (all causes) is approximately 3 per 100,000 per year. Acquired central DI secondary to pituitary surgery is the most common clinical context, occurring transiently in up to 30–50% of patients undergoing transsphenoidal surgery for pituitary adenoma and permanently in 1–5%.
- Nephrogenic DI: X-linked nephrogenic DI (AVPR2 mutations) affects approximately 1 in 250,000 male births — the most common hereditary form. Autosomal recessive AQP2-mutation DI is substantially rarer. Acquired nephrogenic DI (lithium-induced being the most common) is far more prevalent than hereditary forms and affects 20–40% of long-term lithium users.
- Primary polydipsia: The most common cause of polyuria/polydipsia in some psychiatric institutions, affecting 20–30% of patients with chronic schizophrenia or other psychotic disorders. Also associated with sarcoidosis, hypothalamic lesions, and idiopathic causes.
- Gestational DI: Occurs in approximately 1–2 per 100,000 pregnancies; often transient and resolves within weeks of delivery. May unmask subclinical central DI or nephrogenic DI during pregnancy.
- Sex distribution: Hereditary nephrogenic DI (AVPR2) is X-linked recessive and therefore predominantly affects males. Central DI and primary polydipsia have no strong sex predilection. Gestational DI affects only pregnant individuals.
3. Pathophysiology
Water homeostasis depends on the precise integration of AVP secretion, renal AVP responsiveness (mediated by aquaporin-2 water channels), and the thirst response — all regulated by the hypothalamic osmoreceptors and the renin-angiotensin-aldosterone system.
Normal AVP Physiology
Arginine vasopressin (AVP; antidiuretic hormone) is a nonapeptide synthesized in the magnocellular neurons of the supraoptic (SON) and paraventricular (PVN) nuclei of the hypothalamus. It is produced as a larger precursor, preprovasopressin, which is co-packaged with its carrier protein neurophysin II and the C-terminal glycopeptide copeptin in neurosecretory granules. The AVP-neurophysin II complex is transported along axons of the hypothalamo-neurohypophyseal tract to nerve terminals in the posterior pituitary (neurohypophysis), where it is stored in secretory granules and released in response to osmotic and non-osmotic stimuli.
Osmotic regulation: Plasma osmolality is the primary regulator of AVP secretion. Osmoreceptors — principally located in the anterior hypothalamus (organum vasculosum of the lamina terminalis, OVLT; subfornical organ, SFO) — detect a rise in effective plasma osmolality (predominantly sodium) and signal the SON and PVN to increase AVP secretion. The osmotic threshold for AVP release is approximately 280 mOsm/kg in normal adults; above this level, AVP rises linearly with osmolality. A 1% rise in plasma osmolality is sufficient to stimulate AVP release.
Non-osmotic regulation: Hypovolemia (greater than 10% reduction in effective circulating volume) and hypotension potently stimulate AVP secretion via baroreceptor input (carotid sinus, aortic arch, cardiac atria) relayed through the nucleus tractus solitarius. Nausea, pain, surgical stress, and angiotensin II also stimulate AVP release. Non-osmotic stimuli can override osmotic suppression of AVP (as in volume-depleted hyponatremia).
Aquaporin-2 and Renal AVP Action
AVP exerts its antidiuretic effect primarily on the collecting duct principal cells through a precisely regulated signaling cascade:
- AVP binds to the V2 receptor (AVPR2) — a Gs-coupled GPCR — on the basolateral membrane of collecting duct principal cells.
- V2R coupling activates adenylyl cyclase, generating cyclic AMP (cAMP) from ATP.
- cAMP activates protein kinase A (PKA), which phosphorylates aquaporin-2 (AQP2) at Ser256 and other sites.
- Phosphorylated AQP2 monomers, which normally reside in cytoplasmic vesicles, undergo exocytosis and insert into the apical (luminal) membrane of principal cells — dramatically increasing water permeability.
- Water moves osmotically from the dilute tubular lumen into the hypertonic medullary interstitium through the apical AQP2 and then across the basolateral membrane via constitutively expressed AQP3 and AQP4, ultimately entering the vasa recta capillaries.
- The result is water reabsorption, concentrated urine (up to 1200 mOsm/kg), and reduced urine volume.
AVP also acts on V1b receptors in the anterior pituitary (stimulating ACTH release) and V1a receptors in vascular smooth muscle (vasoconstriction at high concentrations). Renal tubular V2R also mediates urea transport through UT-A1/UT-A3 in the inner medullary collecting duct, contributing to the corticomedullary osmotic gradient essential for urinary concentration.
Central DI Pathophysiology
Central DI results from destruction, damage, or dysfunction of the magnocellular neurons in the SON/PVN or their axonal projections to the posterior pituitary. Because the hypothalamic nuclei have reserve capacity, clinical polyuria typically manifests only when greater than 80–90% of AVP-secreting neurons are destroyed. Hence, even large pituitary macroadenomas rarely cause central DI — the posterior pituitary continues to function because the hypothalamic cell bodies are intact. Instead, central DI follows lesions higher in the hypothalamic-neurohypophyseal axis (hypothalamic tumors, infiltrative diseases, trauma to the pituitary stalk).
The "triple phase" response classically follows pituitary stalk transection or severe head injury:
- Phase 1 (0–5 days): Immediate DI from inhibition of axonal AVP release due to neuronal dysfunction (axon shock). Polyuria and dilute urine.
- Phase 2 (5–14 days): Uncontrolled AVP release from degenerating axonal terminals (lysed neurons release stored AVP); may cause transient SIADH with hyponatremia. Urine becomes concentrated inappropriately.
- Phase 3 (beyond 14 days): Permanent central DI if sufficient AVP-secreting neurons are destroyed. The SIADH phase does not invariably occur; the third phase may represent partial or complete permanent DI.
Nephrogenic DI Pathophysiology
In nephrogenic DI, AVP secretion is intact but the collecting duct fails to respond. The defect may be at the level of:
- AVPR2 (V2 receptor): Loss-of-function mutations in the X-linked gene AVPR2 account for 90% of hereditary nephrogenic DI. Greater than 300 distinct mutations are described (missense, frameshift, nonsense, splice-site). The resulting receptor protein fails to reach the plasma membrane (misfolding, ER retention) or cannot couple to Gs. Males with hemizygous mutations are typically fully affected; carrier females show variable expression due to X-inactivation patterns.
- Aquaporin-2 (AQP2): Autosomal recessive (or rarely dominant) mutations in AQP2 account for 10% of hereditary nephrogenic DI. Dominant mutations typically affect AQP2 trafficking (interaction with AQP3/AQP4 at the basolateral membrane or with sorting proteins), while recessive mutations disrupt AQP2 folding, oligomerization, or retention in ER.
- Acquired nephrogenic DI mechanisms:
- Lithium: Li⁺ enters principal cells via the epithelial sodium channel (ENaC) and inhibits glycogen synthase kinase-3β (GSK-3β) and adenylyl cyclase, impairing cAMP generation and AQP2 phosphorylation. Long-term lithium exposure also reduces AQP2 and AQP3 protein expression through downregulation of V2R signaling. Lithium-induced nephrogenic DI may be partially or fully reversible with discontinuation, but in some patients permanent structural tubular damage occurs.
- Hypercalcemia: Calcium activates CaSR in the thick ascending limb, inhibiting NKCC2 and the medullary countercurrent mechanism; calcium also directly inhibits adenylyl cyclase V2R signaling in collecting duct cells and promotes AQP2 internalization.
- Hypokalemia: Chronic hypokalemia reduces AQP2 expression and impairs the corticomedullary osmotic gradient.
- Chronic tubulointerstitial nephritis: Direct destruction of collecting duct architecture impairs AVP-mediated water reabsorption.
- Osmotic diuresis: Glucose, urea, or mannitol dilute the medullary interstitium and impair urinary concentration independently of AVP signaling.
Dipsogenic DI (Primary Polydipsia)
In primary polydipsia, the primary disturbance is excessive water intake — driven by a pathologically lowered osmotic threshold for thirst (dipsogenic form), habit or compulsion (psychogenic polydipsia in psychiatric patients), or hypothalamic damage altering thirst perception. Excessive water intake suppresses plasma osmolality below the normal AVP secretion threshold, inhibiting AVP release and producing physiologically appropriate dilute polyuria. Over time, the renal medullary gradient is "washed out," and the kidney loses some concentrating ability — complicating distinction from central DI. The kidneys remain capable of responding to exogenous desmopressin, but administering it in primary polydipsia causes dangerous water retention and hyponatremia.
4. Etiology and Risk Factors
Central Diabetes Insipidus
- Idiopathic: Accounts for 30–50% of central DI in adults. Imaging may be normal; anti-AVP (anti-neurophysin II) autoantibodies are detectable in up to 30–50%, suggesting autoimmune destruction of AVP neurons. MRI typically shows loss of the posterior pituitary T1-hyperintense "bright spot."
- Post-surgical: Transsphenoidal pituitary surgery (most common surgical cause), craniopharyngioma resection, hypothalamic tumor surgery. Permanent postoperative central DI develops in 1–5% of standard pituitary adenoma surgeries and in 30–60% of craniopharyngioma surgeries.
- Traumatic brain injury: Transient or permanent central DI following severe TBI; incidence approximately 15–20% in critical care settings. Hypoxic-ischemic injury to the hypothalamus/pituitary stalk from hypotension or basilar skull fractures is the mechanism.
- Tumors:
- Craniopharyngioma (children and young adults): most common tumor causing central DI in pediatrics
- Germinoma (hypothalamic/suprasellar): highly associated with central DI — may present before tumor is visible on MRI
- Pituitary metastases (breast, lung): unusual but DI is the presenting manifestation in 75% of posterior pituitary metastases
- Langerhans cell histiocytosis (LCH): central DI in 15–25% of patients with systemic LCH; characteristic pituitary stalk thickening on MRI
- Lymphoma, germ cell tumors
- Infiltrative and granulomatous diseases: Neurosarcoidosis, IgG4-related disease (infundibulo-hypophysitis), lymphocytic infundibulo-neurohypophysitis, Wegener granulomatosis, tuberculosis — all can infiltrate the pituitary stalk and hypothalamus.
- Autoimmune: Lymphocytic hypophysitis (particularly affecting the neurohypophysis/infundibulum); strong association with immune checkpoint inhibitor (ICI) therapy (ipilimumab, nivolumab) — a recognized immune-related adverse event in cancer therapy.
- Vascular: Sheehan syndrome (postpartum pituitary infarction), cerebrovascular accident involving the hypothalamic-pituitary axis, Wernicke encephalopathy.
- Hereditary (familial central DI): Autosomal dominant mutations in the AVP gene (AVP-NPII gene); characterized by progressive postnatal destruction of AVP-secreting neurons, with DI typically manifesting in childhood rather than at birth. The mutant preprovasopressin misfolds in the ER, triggering ER stress and progressive neuronal apoptosis.
- Wolfram syndrome (DIDMOAD): Autosomal recessive mutations in WFS1 (wolframin); DI with Diabetes mellitus, Optic Atrophy, and Deafness. Rare but important to recognize in young patients.
Nephrogenic Diabetes Insipidus
- X-linked (AVPR2 mutations): Most common hereditary form; presents in neonates with hypernatremia, failure to thrive, fever, and dilute urine despite hyperosmolality.
- Autosomal recessive/dominant (AQP2 mutations): Rarer; variable severity depending on mutation type and zygosity.
- Lithium toxicity: Most common cause of acquired nephrogenic DI; affects 20–40% of long-term lithium users.
- Hypercalcemia: Causes nephrogenic DI when serum calcium is persistently elevated (greater than 11 mg/dL).
- Hypokalemia: Chronic potassium depletion (less than 3 mEq/L, particularly with prolonged depletion).
- Medications: Demeclocycline (historically used to treat SIADH — it blocks V2R signaling); amphotericin B; foscarnet; cidofovir; ifosfamide; colchicine.
- Chronic kidney disease: Tubulointerstitial diseases, obstructive uropathy, sickle cell nephropathy, Sjögren syndrome with renal tubular involvement.
- Infiltrative renal diseases: Amyloidosis, sarcoidosis of the kidney.
Primary Polydipsia Risk Factors
- Psychiatric disorders: schizophrenia (20–30% prevalence of polydipsia), schizoaffective disorder, major depression, OCD
- Psychotropic medications stimulating thirst or causing dry mouth: anticholinergics, clozapine, carbamazepine
- Hypothalamic lesions altering thirst osmostatic control (dipsogenic subtype)
- Behavioral/habitual polydipsia (athletes, health-conscious individuals)
5. Clinical Presentation
Core Symptoms
- Polyuria: The cardinal symptom; urine output typically 3–20 liters per day (normal: 1–2.5 L/day). Complete central or complete nephrogenic DI produces maximal urine output of up to 18–20 L/day. Partial DI produces less severe polyuria.
- Polydipsia: Extreme thirst driving compensatory fluid intake. In patients with intact thirst and free access to water, plasma osmolality and sodium remain near normal despite massive fluid turnover. Patients with hypothalamic damage impairing both AVP secretion and thirst perception (adipsic/hypodipsic DI) are at highest risk for severe hypernatremia and death.
- Nocturia: Polyuria is not limited to daytime; nocturia is prominent and often the symptom that first prompts medical attention.
- Preference for cold water: Characteristic of DI (particularly central DI), in contrast to primary polydipsia patients who often drink large volumes at variable temperatures.
- Dilute urine: Patients may observe pale, nearly colorless urine.
Age-Specific Presentations
- Neonates and infants: Congenital nephrogenic DI presents in male neonates with hypernatremic dehydration, fever, irritability, failure to thrive, vomiting, and feeding difficulties. The urine is inappropriately dilute (urine osmolality less than 200 mOsm/kg) despite markedly elevated serum osmolality and sodium. Cerebral injury and cognitive impairment result from repeated hypernatremic episodes.
- Children: Enuresis, secondary nocturnal enuresis, school performance difficulties from sleep disruption, growth retardation (from poor caloric intake due to excessive water consumption displacing food). Hydronephrosis from bladder overdistension.
- Adults: Predominantly polyuria and polydipsia. Work and sleep disruption. In post-surgical or post-traumatic central DI, the triple-phase response may produce confusing fluctuations in urine output and sodium.
Signs of Dehydration and Hypernatremia
When fluid intake cannot keep pace with urinary losses (impaired consciousness, restricted access to water, impaired thirst):
- Dry mucous membranes, decreased skin turgor, orthostatic hypotension, tachycardia
- Hypernatremia: serum sodium greater than 145 mEq/L, rising to greater than 160 mEq/L in severe cases
- Neurologic manifestations of hypernatremia: irritability, confusion, lethargy, seizures (from brain cell shrinkage and pontine/cortical vein rupture in severe, rapid hypernatremia)
- Rhabdomyolysis: rare complication of severe hypernatremia
Central DI-Specific Features
- Symptoms may begin acutely (post-surgical or post-traumatic) or insidiously (infiltrative, autoimmune, hereditary)
- Associated features of anterior pituitary hormone deficiencies (fatigue, amenorrhea, growth failure, hypothyroid symptoms) when the pituitary stalk or anterior pituitary is also damaged
- Visual field defects if a suprasellar mass (craniopharyngioma, germinoma) is responsible
- MRI finding: loss of posterior pituitary T1-bright spot (normally visible as a hyperintense signal due to lipid-rich AVP secretory granules); pituitary stalk thickening (greater than 3 mm) in infiltrative causes
Nephrogenic DI-Specific Features
- Congenital X-linked form: neonatal presentation in males; carrier mothers may have mild or no symptoms
- Acquired: onset correlates with duration of lithium use or degree of hypercalcemia
- Dilated urinary tract (hydronephrosis, megaureter, megacystis) from chronic high urine volumes in children with untreated congenital NDI
6. Diagnosis
The diagnostic workup proceeds in two stages: (1) confirming that the patient has DI (as opposed to primary polydipsia) and (2) determining the subtype (central vs. nephrogenic).
Initial Evaluation
- 24-hour urine volume: Polyuria defined as urine output greater than 3 L/day in adults (greater than 2 L/m²/day in children). DI typically produces 4–18 L/day; primary polydipsia may produce 4–10 L/day.
- Serum sodium and osmolality: Simultaneous measurement is essential. Normal or high-normal sodium (greater than 142 mEq/L) with polyuria strongly favors DI; low-normal sodium (less than 138 mEq/L) with polyuria favors primary polydipsia.
- Urine osmolality (spot): Values less than 300 mOsm/kg are consistent with DI; values greater than 800 mOsm/kg essentially exclude DI. The simultaneous relationship between serum and urine osmolality is critical.
- Serum biochemistry: Glucose (to exclude osmotic diuresis from hyperglycemia), calcium (exclude hypercalcemia-induced nephrogenic DI), potassium, BUN, creatinine.
- Plasma AVP: Low or undetectable in central DI; elevated in nephrogenic DI. However, plasma AVP assay is technically challenging (rapid degradation, platelet contamination); largely supplanted by copeptin measurement in centers with access.
Water Deprivation Test
The water deprivation test (Fasting Test; Miller test) is the traditional diagnostic standard. It exploits the fact that dehydration should maximally stimulate AVP secretion and urinary concentration in a normal individual, whereas DI patients cannot concentrate urine appropriately.
Protocol:
- Fluid restriction from midnight (or from 8 AM in a supervised clinical setting); patient weighed hourly. Test is discontinued if body weight drops more than 3–5% (excessive dehydration).
- Urine osmolality, urine specific gravity, plasma osmolality, and serum sodium measured hourly.
- Dehydration continues until urine osmolality reaches a plateau (less than 30 mOsm/kg change in three consecutive measurements) or plasma osmolality exceeds 295–300 mOsm/kg or serum sodium exceeds 146 mEq/L.
- Desmopressin response phase: At the plateau, desmopressin 1–2 mcg SC or IV (or 10–20 mcg intranasal) is administered. Urine and plasma osmolality are measured at 30, 60, and 120 minutes post-administration.
Interpretation:
- Normal / Primary polydipsia: During dehydration, urine osmolality rises above 800 mOsm/kg (normal response); desmopressin produces less than 10% further increase (AVP axis is already maximally stimulated).
- Complete central DI: Urine osmolality remains below 300 mOsm/kg despite dehydration; desmopressin produces a dramatic increase in urine osmolality of greater than 50% (often to 400–800 mOsm/kg or higher), confirming preserved V2R/AQP2 responsiveness.
- Partial central DI: Urine osmolality rises to 300–800 mOsm/kg with dehydration; desmopressin produces 10–50% further increase.
- Complete nephrogenic DI: Urine osmolality remains below 300 mOsm/kg during dehydration AND shows less than 10% increase after desmopressin administration (V2R or AQP2 defect precludes response).
- Partial nephrogenic DI: Urine osmolality rises modestly during dehydration; desmopressin produces less than 10–50% increase, reflecting residual V2R function or partial AQP2 defect.
Limitations: The water deprivation test has significant diagnostic accuracy limitations — particularly in the partial forms — with sensitivity and specificity for correctly classifying central vs. nephrogenic DI vs. primary polydipsia of only 70–80%. Misclassification is most common for partial central DI and primary polydipsia. The test requires careful supervision to avoid dangerous hypernatremia.
Copeptin-Based Diagnostic Protocol
Copeptin is the C-terminal portion of the preprovasopressin precursor molecule, secreted equimolarly with AVP. It is stable at room temperature for up to 7 days, accurately measured by commercial immunoassays, and reflects AVP secretion with high fidelity — overcoming the technical limitations of direct AVP measurement.
Hypertonic saline stimulation test (Fenske/Christ-Crain protocol): Hypertonic saline (3% NaCl) is infused intravenously at 0.15 mL/kg/min until plasma sodium reaches 150 mEq/L or copeptin is measured when plasma sodium reaches 147–148 mEq/L.
- Copeptin less than 2.6 pmol/L: confirms complete central DI
- Copeptin greater than 4.9 pmol/L during osmotic stimulation: confirms nephrogenic DI or primary polydipsia (AVP axis intact)
- Copeptin 2.6–4.9 pmol/L: indeterminate; may reflect partial central DI
The hypertonic saline-stimulated copeptin test demonstrated 96% diagnostic accuracy for differentiating central DI from primary polydipsia in the pivotal trial by Fenske et al. (NEJM 2018), compared to 76% for the water deprivation test in the same cohort.
Arginine-stimulated copeptin: IV arginine (30 g infused over 30 minutes) stimulates copeptin release via non-osmotic mechanisms. A copeptin level greater than 3.8 pmol/L at peak distinguishes primary polydipsia from central DI with high accuracy, without the risks of hypertonic saline. Validated in two prospective trials; a copeptin-based algorithm using arginine stimulation is now recommended as first-line by the European Society of Endocrinology (2022 guidelines) when copeptin measurement is available.
Differentiating Central from Nephrogenic DI
- Baseline plasma copeptin: Markedly elevated (greater than 21.4 pmol/L) indicates nephrogenic DI (AVP secretion is high but ineffective); this threshold alone diagnoses nephrogenic DI without requiring a stimulation test.
- Desmopressin response: As above — dramatic urine concentration response confirms central DI; absent response confirms nephrogenic DI.
- Genetic testing: For suspected hereditary nephrogenic DI — sequencing of AVPR2 (X-linked) and AQP2 (autosomal) is diagnostic in familial or neonatal cases.
- MRI brain/pituitary: Essential in central DI — loss of posterior pituitary T1 bright spot, pituitary stalk thickening (greater than 3.5 mm), suprasellar mass, or hypothalamic lesion guides etiologic workup. Normal MRI does not exclude central DI (idiopathic autoimmune forms may have subtle findings).
Etiologic Workup
Once central DI is confirmed, systematic etiologic evaluation includes:
- MRI pituitary/hypothalamus with and without gadolinium (FIESTA or other high-resolution sequences for stalk assessment)
- Anti-AVP or anti-neurophysin antibodies (research assays; not widely available)
- Serum and urine protein electrophoresis, free light chains (myeloma, amyloidosis)
- ACE level, calcium, chest CT (sarcoidosis)
- CSF beta-hCG and AFP (germinoma)
- IgG4 level (IgG4-related hypophysitis)
- LDH, peripheral smear (lymphoma)
- Langerhans cell histiocytosis evaluation: skin biopsy, BRAF V600E mutation testing (in suspected LCH)
- Anterior pituitary function testing: free T4, TSH, cortisol, IGF-1, LH/FSH, testosterone or estradiol (pan-pituitary involvement is common)
7. Treatment
Central and Gestational Diabetes Insipidus
Desmopressin (DDAVP; 1-deamino-8-D-arginine vasopressin): A synthetic AVP analog with selective V2R agonism (minimal V1 activity; no pressor effect), prolonged half-life (8–24 hours), and resistance to degradation by vasopressinase. It is the treatment of choice for central DI and gestational DI.
- Intranasal desmopressin: Metered nasal spray (10 mcg/spray) or rhinyl tube (100 mcg/mL). Bioavailability 3–5%. Dose: 10–20 mcg once or twice daily, individualized. Nasal congestion, rhinitis, or surgery can alter absorption significantly.
- Oral desmopressin (tablet or oral lyophilisate): Bioavailability 0.5–1%. Tablet doses: 0.1–0.4 mg two to three times daily. Oral lyophilisate (Nocdurna, Noctiva): 25–50 mcg sublingually, primarily used for nocturia; FDA-approved for adult-onset nocturia from nocturnal polyuria.
- Parenteral desmopressin: IV or SC 1–2 mcg per dose; 10-fold more potent than intranasal. Used perioperatively, in hospitalized patients, and in hypercalcemic crisis.
- Dosing strategy: Begin with the lowest effective dose; allow a brief period of mild polyuria between doses (a "escape window") to prevent cumulative water retention and hyponatremia. Patients should be educated to reduce fluid intake when taking desmopressin and to hold a dose if sodium is falling or they have unusual fluid retention.
Key safety issue — hyponatremia: The most serious complication of desmopressin therapy is iatrogenic hyponatremia from excessive water intake in the presence of pharmacologically maintained antidiuresis. Symptoms range from mild (nausea, headache) to severe (seizures, respiratory arrest, death). Patient education on maintaining appropriate fluid intake is essential. Sodium monitoring at baseline, at 1 week, and periodically thereafter is recommended.
Gestational DI: Desmopressin is the treatment of choice; it is not degraded by placental vasopressinase (unlike AVP) and does not stimulate uterine oxytocin receptors at antidiuretic doses. It crosses the placenta minimally and is classified as pregnancy category B. Typically resolves within 4–6 weeks postpartum.
Nephrogenic Diabetes Insipidus
Treatment of nephrogenic DI is directed at the underlying cause when reversible (lithium discontinuation, correction of hypercalcemia or hypokalemia) and symptomatic reduction of polyuria through paradoxical antidiuretics when the defect is irreversible.
- Adequate hydration: In young children with congenital NDI, adequate hypotonic fluid intake is the most critical intervention to prevent hypernatremic dehydration and cognitive damage. Nasogastric tube feeding may be required in infants.
- Low-sodium, low-protein diet: Reduces the obligate solute load requiring urinary excretion, decreasing total urine volume. Dietary sodium restriction to 0.5–1.5 mEq/kg/day in children with congenital NDI is a standard adjunct.
- Thiazide diuretics (hydrochlorothiazide): Paradoxically reduce urine volume in DI by inducing mild volume contraction. Volume depletion activates proximal tubular sodium and water reabsorption (through the isotonic proximal tubule), reducing distal delivery of filtrate to the AVP-insensitive collecting duct. Dose in children: 1–2 mg/kg/day. Combination with amiloride (which blocks ENaC-mediated lithium uptake in principal cells) is particularly effective for lithium-induced NDI and reduces lithium-induced potassium wasting.
- Indomethacin (NSAID): Reduces prostaglandin-mediated inhibition of AVP action in the collecting duct; reduces urine volume by 30–50% when combined with thiazide diuretics in congenital NDI. Risk: gastrointestinal ulceration, reduced renal blood flow; generally limited to patients with severe disease not controlled by thiazides alone.
- High-dose desmopressin: In patients with partial nephrogenic DI (AVPR2 mutations with some residual receptor function or AQP2 mutations affecting trafficking but not ligand binding), pharmacologic doses of desmopressin may partially overcome the defect. Most complete nephrogenic DI patients do not respond.
- Lithium-induced NDI management:
- Discontinue lithium if clinically feasible (in consultation with psychiatry).
- If lithium must be continued: switch to once-daily lithium dosing (reduces tubular exposure), use amiloride (which blocks lithium entry into collecting duct cells via ENaC), maintain serum lithium levels in the lower therapeutic range.
- After lithium discontinuation, NDI may partially improve over weeks to months; complete resolution is uncommon after years of exposure due to structural tubular damage.
Primary Polydipsia
- Treat the underlying psychiatric disorder; atypical antipsychotics may be helpful in psychogenic polydipsia in schizophrenia.
- Behavioral interventions: supervised fluid restriction with behavioral therapy; close monitoring to prevent hyponatremia in psychiatric inpatients.
- Desmopressin is contraindicated in primary polydipsia — it will exacerbate water retention and cause life-threatening hyponatremia.
Management of Hypernatremia in DI
- Acute severe hypernatremia (greater than 160 mEq/L): correct gradually using hypotonic fluids; rate of sodium correction should not exceed 10–12 mEq/L per 24 hours (or 0.5 mEq/L per hour) to prevent cerebral edema from rapid osmotic shift. Free water deficit = TBW × (serum Na − 140) / 140.
- Chronic hypernatremia (developed slowly over more than 48 hours): correct even more gradually (8–10 mEq/L per day) due to cerebral adaptive idiogenic osmole accumulation.
- Administer desmopressin IV or SC simultaneously with fluid repletion to reduce ongoing urinary losses in central DI.
Perioperative Management
- Patients with central DI undergoing surgery should have IV access for desmopressin administration when NPO.
- Careful fluid balance and hourly urine output monitoring postoperatively; be alert to the triple-phase response after pituitary or hypothalamic surgery.
- Plasma sodium should be measured every 4–6 hours in the immediate postoperative period following transsphenoidal surgery.
8. Complications
From Untreated or Undertreated DI
- Hypernatremia and hypernatremic dehydration: The most immediate and life-threatening complication, particularly in infants, the elderly, and patients with impaired thirst or consciousness. Can cause seizures, intracranial hemorrhage (from brain shrinkage and dural sinus stretching), permanent neurologic injury, and death.
- Nephrogenic complications of chronic polyuria: Bladder overdistension, hydroureter, hydronephrosis, megacystis — particularly in children with congenital NDI. Obstructive nephropathy from these structural changes may itself impair renal concentrating ability further and progress to CKD.
- Growth retardation and cognitive impairment (children): Recurrent hypernatremic episodes during early brain development cause intellectual disability and developmental delay. The severity of cognitive impairment correlates with the number and severity of hypernatremic episodes in early childhood.
- Nutritional complications: Infants with NDI fill their limited gastric capacity with water rather than milk, leading to caloric deficiency and severe failure to thrive. Persistent vomiting from excessive fluid intake compounds this.
From Treatment (Desmopressin)
- Hyponatremia: The most clinically important treatment complication. Can be severe (sodium less than 125 mEq/L) and rapidly symptomatic. Most commonly occurs when patients continue high fluid intake during desmopressin therapy, during intercurrent illness with inappropriate fluid administration, or when patients take excess desmopressin doses.
- Cerebral edema: Rapid hyponatremia from desmopressin therapy may cause brain swelling, particularly in children and pre-menopausal women who have less efficient brain volume adaptation mechanisms.
- Angina and allergic reactions: Rare, primarily with parenteral formulations.
- Nasal irritation and epistaxis: From intranasal desmopressin formulations.
Complications of Underlying Cause
- Adipsic/hypodipsic DI (combined thirst and AVP deficiency from hypothalamic lesions): Among the most dangerous clinical scenarios; patients cannot autoregulate fluid balance. Severe, recurrent hypernatremia alternating with iatrogenic hyponatremia. Requires an extremely structured fluid intake protocol and meticulous sodium monitoring.
- Panhypopituitarism from pituitary stalk damage affecting all anterior pituitary hormones simultaneously with central DI.
- Cognitive and emotional effects of chronic disease management burden in children and families.
9. Prognosis
- Central DI: With appropriate desmopressin therapy and patient education, patients with central DI lead normal lives with excellent quality of life. Prognosis is primarily determined by the underlying etiology — idiopathic central DI has an excellent outlook; traumatic or post-surgical DI may be permanent but manageable; central DI from malignancy (germinoma, metastases) carries the prognosis of the underlying cancer.
- Congenital nephrogenic DI: Historically associated with significant cognitive impairment from repeated hypernatremic episodes; early diagnosis (neonatal genetic testing in families at risk) and aggressive prevention of hypernatremia substantially improve neurologic outcomes. With optimal management, intellectual function can be preserved. Structural urinary tract complications (hydronephrosis) may require urologic intervention.
- Acquired nephrogenic DI: Lithium-induced NDI partially reverses after lithium discontinuation in most patients; complete reversal is uncommon after decades of therapy. Hypercalcemia-induced NDI resolves with successful treatment of hypercalcemia.
- Adipsic DI: A particularly challenging form with high morbidity from alternating severe dysnatremia; standardized fluid prescription protocols guided by body weight and daily sodium monitoring are the cornerstone of management. Mortality is substantially higher than for other forms of DI.
- Post-surgical (transient) central DI: The majority of post-transsphenoidal cases resolve within days to weeks. Permanent DI is less common and requires lifelong desmopressin therapy.
10. Prevention
- Monitoring during lithium therapy: Baseline renal function, urine osmolality, and serum sodium before initiation; annual monitoring of creatinine, eGFR, and urine concentration ability during chronic therapy. Use the lowest effective lithium dose; consider once-daily dosing to minimize tubular exposure. Amiloride co-administration is nephroprotective in patients developing NDI who must continue lithium.
- Genetic counseling: Families with known AVPR2 or AQP2 mutations should receive genetic counseling and carrier testing before and during pregnancy. Prenatal genetic diagnosis or preimplantation genetic testing is available. Newborn males born to carrier mothers should be tested immediately.
- Neonatal screening (at-risk families): Measurement of plasma sodium and urine osmolality in the first days of life in males at risk for X-linked NDI enables diagnosis before the first hypernatremic crisis.
- Peri-operative planning: Anticipate transient or permanent DI in patients undergoing transsphenoidal pituitary surgery or hypothalamic tumor resection; arrange postoperative sodium monitoring protocols and desmopressin availability.
- Prompt treatment of hypercalcemia and hypokalemia to prevent acquired NDI.
- Immune checkpoint inhibitor monitoring: Patients receiving anti-CTLA4 (ipilimumab) or anti-PD1/PDL1 agents should be monitored for endocrine immune-related adverse events including hypophysitis and central DI — baseline and periodic pituitary hormone testing and awareness of DI symptoms.
- Education of patients and caregivers: Patients on desmopressin must understand the risk of hyponatremia from excessive fluid intake and be instructed on appropriate fluid management, sick-day rules, and when to seek medical attention.
11. Recent Research
- Copeptin-based diagnostic algorithms (ESE 2022 Guidelines): The 2022 European Society of Endocrinology Clinical Practice Guideline formally recommends copeptin measurement as the preferred first-line diagnostic tool, with arginine-stimulated copeptin testing proposed for the initial evaluation and hypertonic saline-stimulated copeptin as the most accurate definitive test. The traditional water deprivation test is recommended only when copeptin is unavailable. This represents a paradigm shift in DI diagnosis.
- Pharmacological chaperones for nephrogenic DI: Misfolded AVPR2 proteins (most hereditary NDI mutations produce ER-retained, structurally intact but misfolded receptors rather than truncated proteins) can be rescued by small-molecule pharmacological chaperones that stabilize the receptor and promote trafficking to the plasma membrane. Non-peptide V2R antagonists (vaptans) paradoxically act as pharmacological chaperones in vitro at sub-pharmacologic concentrations. Clinical trials of chaperone therapy (tolvaptan at sub-antagonist doses) for selected AVPR2 mutations are ongoing.
- Gene therapy for congenital NDI: AAV-mediated delivery of functional AVPR2 or AQP2 to renal tubular cells has been demonstrated in mouse models of NDI; clinical translation is in early development. An alternative approach delivers AQP2 to the urinary epithelium via intravesical gene therapy (targeting the bladder to restore some capacity for water reabsorption from the bladder wall).
- AQP2 trafficking pharmacology: The cAMP-independent AQP2 trafficking pathway — stimulated by calcitonin (CTb receptor/cAMP), prostaglandin E2 (EP4/cAMP), and the NO-cGMP pathway — offers alternative drug targets for nephrogenic DI. Roflumilast (PDE4 inhibitor), cilostazol (PDE3 inhibitor), and metformin (AMPK activation, independent of cAMP) have demonstrated AQP2 membrane insertion in preclinical models.
- Subcutaneous desmopressin formulations: A once-daily subcutaneous desmopressin formulation (Nocdurna SC, under investigation) provides more consistent pharmacokinetics than intranasal delivery, potentially reducing hyponatremia risk through more predictable dosing. Phase 3 trials are ongoing.
- Adipsic DI management protocols: Structured algorithm-based fluid management systems, in which fluid intake is prescribed based on daily body weight changes and intermittent serum sodium monitoring, have demonstrated improved sodium stability and quality of life in adipsic DI cohorts at specialized pituitary centers.
- Long-term cognitive outcomes in neonatal NDI: Longitudinal follow-up studies confirm that children diagnosed early through neonatal screening and managed to prevent hypernatremic episodes achieve near-normal cognitive and educational outcomes — strongly supporting newborn screening in at-risk families.
12. References
- Fenske W, Refardt J, Chifu I, et al. A copeptin-based approach in the diagnosis of diabetes insipidus. N Engl J Med. 2018;379(5):428-439.
- Christ-Crain M, Bichet DG, Fenske WK, et al. Diabetes insipidus. Nat Rev Dis Primers. 2019;5(1):54.
- Refardt J, Winzeler B, Christ-Crain M. Diabetes insipidus: an update. Endocrinol Metab Clin North Am. 2020;49(3):517-531.
- Robertson GL. Diabetes insipidus: differential diagnosis and management. Best Pract Res Clin Endocrinol Metab. 2016;30(2):205-218.
- Bockenhauer D, Bichet DG. Pathophysiology, diagnosis and management of nephrogenic diabetes insipidus. Nat Rev Nephrol. 2015;11(10):576-588.
- Chitturi S, Harris M, Van Daal A, Brown C, Henderson S. Diabetes insipidus following pituitary surgery: incidence and predictors. Pituitary. 2016;19(4):347-354.
- Majzoub JA, Muglia LJ. Molecular medicine. Knockout mice. N Engl J Med. 1996;334(14):904-907.
- Morello JP, Bichet DG. Nephrogenic diabetes insipidus. Annu Rev Physiol. 2001;63:607-630.
- Trepiccione F, Christensen BM. Lithium-induced nephrogenic diabetes insipidus: new clinical and experimental findings. J Nephrol. 2010;23(Suppl 16):S43-S48. PMID:21170886
- Grünfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol. 2009;5(5):270-276.
- Bichet DG. Vasopressin receptor mutations causing nephrogenic diabetes insipidus. Semin Nephrol. 2008;28(3):245-251.
- Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17(4):471-503.
- Timper K, Fenske W, Kühn F, et al. Diagnostic accuracy of copeptin in the differential diagnosis of the polyuria-polydipsia syndrome: a systematic review. J Clin Endocrinol Metab. 2015;100(6):2268-2274.
- Maghnie M, Cosi G, Genovese E, et al. Central diabetes insipidus in children and young adults. N Engl J Med. 2000;343(14):998-1007.
- Christensen JH, Rittig S. Familial neurohypophyseal diabetes insipidus — an update. Semin Nephrol. 2006;26(3):209-223.
- Winzeler B, Cesana-Nigro N, Refardt J, et al. Arginine-stimulated copeptin measurements in the differential diagnosis of diabetes insipidus: a prospective diagnostic study. Lancet. 2019;394(10197):587-595.
- Arima H, Oiso Y, Juul KV, Nørgaard JP. Nonadherence with pharmacological treatment of desmopressin in diabetes insipidus. J Clin Endocrinol Metab. 2012;97(1):6-15.
- Christ-Crain M, Winzeler B, Refardt J. Diagnosis and management of diabetes insipidus for the internist: an update. J Intern Med. 2021;290(1):73-87.
Research Papers
The following PubMed topic searches aggregate the current peer-reviewed literature. Each link opens a live PubMed query — results update as new studies are indexed.
- PubMed — diabetes insipidus
- PubMed — central diabetes insipidus
- PubMed — nephrogenic diabetes insipidus
- PubMed — arginine vasopressin deficiency
- PubMed — desmopressin treatment
- PubMed — water deprivation test
- PubMed — copeptin diagnosis
- PubMed — hypernatremia diabetes insipidus
- PubMed — lithium nephrogenic diabetes insipidus
- PubMed — pituitary posterior lobe
- PubMed — polyuria polydipsia
- PubMed — gestational diabetes insipidus
Connections
- Endocrinology
- Thirst, ADH & How Your Body Guards Its Water — interactive animation
- Diabetes
- Addison's Disease
- Cushing's Syndrome
- Hyperparathyroidism
- Metabolic Syndrome
- Thyroid Disorders
- Gestational Diabetes
- Kidney Disease
- Edema
- Potassium
- Calcium
- Arginine
- eGFR
- Creatinine
- Urinalysis
- Pheochromocytoma