Cardiomyopathy
Interactive Visualization How Heart Failure Develops Weaken the pump and watch the ejection fraction fall, fluid back up into the lungs, and the body's own 'fixes' quietly make it worse — then unload it with the right drugs. Launch →
Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research and Advances
- Research Papers
- Connections
- Featured Videos
1. Overview
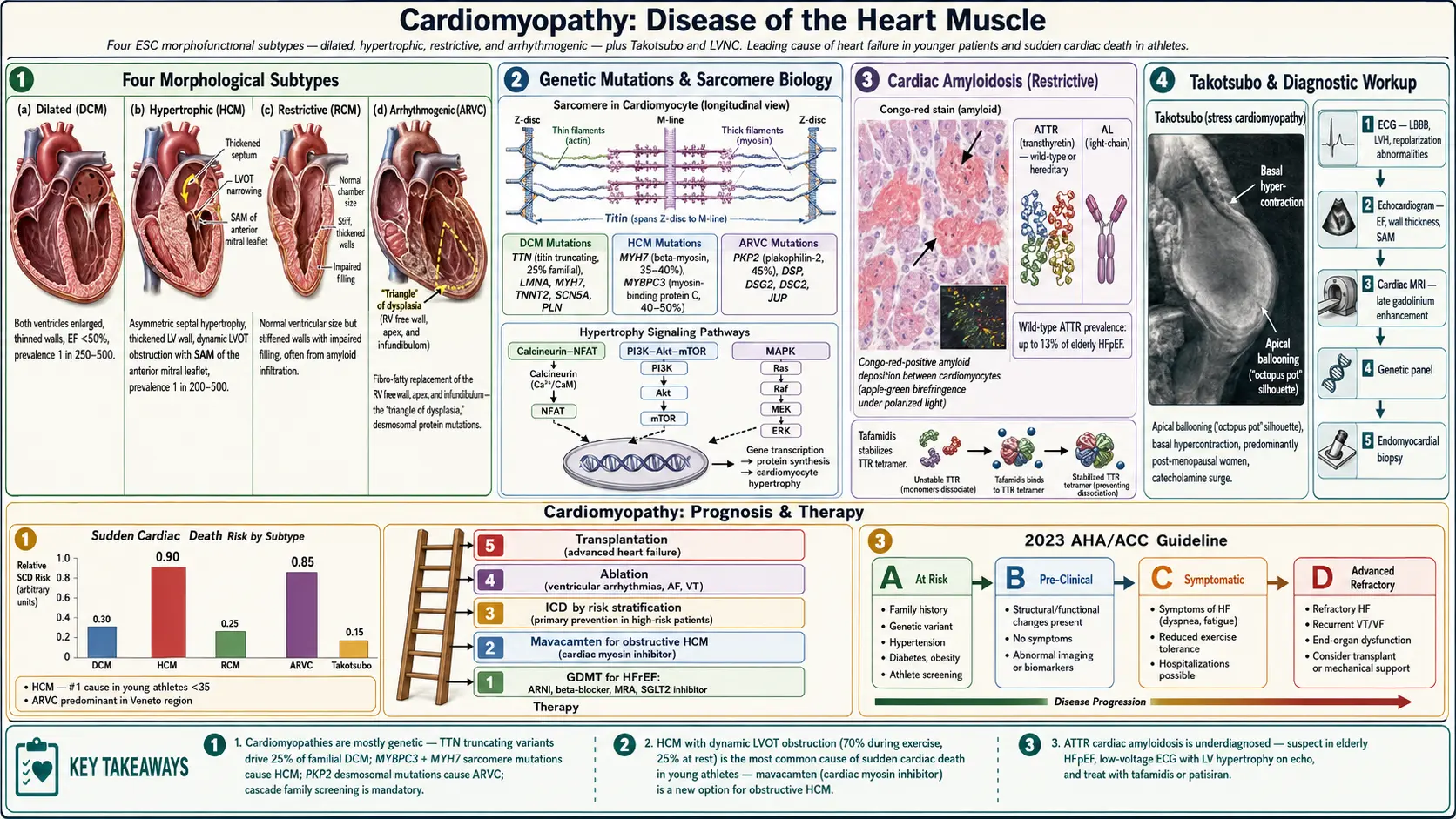
Cardiomyopathy refers to a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation, arising from a variety of causes that are frequently genetic. Cardiomyopathies are the leading cause of heart failure in younger patients and a major cause of sudden cardiac death.
The 2006 AHA classification divides cardiomyopathies into primary (predominantly involving the heart) and secondary (myocardial involvement as part of generalized systemic disease). Primary cardiomyopathies are further classified as genetic, mixed (genetic + non-genetic), and acquired. The 2008 European Society of Cardiology (ESC) classification uses a morphofunctional approach identifying four major subtypes:
- Dilated cardiomyopathy (DCM): Ventricular dilatation with systolic dysfunction
- Hypertrophic cardiomyopathy (HCM): Inappropriate myocardial hypertrophy, usually asymmetric, not explained by loading conditions
- Restrictive cardiomyopathy (RCM): Impaired ventricular filling with preserved or near-normal systolic function
- Arrhythmogenic right ventricular cardiomyopathy (ARVC): Fibro-fatty replacement of right ventricular myocardium
- Unclassified: Left ventricular non-compaction (LVNC), Takotsubo (stress) cardiomyopathy
The 2023 AHA/ACC Guideline on Cardiomyopathies provides the most current framework, introducing the concept of Stage A (at risk) through Stage D (advanced) cardiomyopathy progression for DCM and HCM.
2. Epidemiology
Cardiomyopathies collectively represent a significant burden of cardiovascular disease worldwide:
- Dilated cardiomyopathy (DCM): Most common cardiomyopathy; prevalence approximately 1 in 250–500 in the general population; accounts for 10,000 deaths and 46,000 hospitalizations annually in the US; leading indication for heart transplantation
- Hypertrophic cardiomyopathy (HCM): Most common inherited cardiac disease; prevalence 1 in 200–500 (0.2–0.5%); affects approximately 600,000–750,000 Americans; most common cause of sudden cardiac death in young athletes (<35 years)
- Arrhythmogenic cardiomyopathy (ARVC): Prevalence 1 in 1,000–5,000; predominant cause of sudden cardiac death in young athletes in Italy (Veneto region); male predominance (3:1)
- Restrictive cardiomyopathy: Rare; cardiac amyloidosis (the most common secondary RCM cause) is increasingly recognized — ATTR amyloidosis affects an estimated 300,000 individuals in the US, with wild-type ATTR affecting up to 13% of elderly patients with HFpEF
- Takotsubo cardiomyopathy: Accounts for 1–2% of all acute coronary syndrome presentations; predominantly postmenopausal women (90%); annual US incidence approximately 100,000 cases
3. Pathophysiology
Dilated Cardiomyopathy (DCM)
DCM is characterized by progressive dilatation of one or both ventricles with impaired systolic function (EF <50%). At the molecular level, genetic mutations in sarcomeric, cytoskeletal, and nuclear envelope proteins disrupt force generation, calcium handling, and structural integrity. TTN (titin) truncating variants are the most common cause, accounting for 25% of familial DCM and 18% of sporadic DCM. Other culprit genes include LMNA (lamin A/C), MYH7, TNNT2, SCN5A, and PLN (phospholamban).
Mechanistically, cardiomyocyte loss (apoptosis, necrosis, autophagy) triggers a maladaptive repair response including: reactive fibrosis (TGF-β/SMAD pathway activation), neurohormonal activation (RAAS, sympathetic nervous system upregulation), and ventricular remodeling — progressive dilation and wall thinning — leading to worsening systolic dysfunction in a vicious cycle.
Hypertrophic Cardiomyopathy (HCM)
HCM results from autosomal dominant mutations in sarcomeric protein genes — most commonly MYH7 (beta-myosin heavy chain, 35–40%) and MYBPC3 (myosin-binding protein C, 40–50%). Mutant sarcomeres exhibit increased ATPase activity and calcium sensitivity, leading to hypercontractility, energy depletion, and disordered calcium flux. This triggers compensatory hypertrophy mediated by calcineurin-NFAT, PI3K-Akt-mTOR, and MAPK signaling cascades.
The hallmark histologic features are myocyte hypertrophy, myofibrillar disarray, and interstitial fibrosis. Asymmetric septal hypertrophy (ASH) — particularly of the basal interventricular septum — creates dynamic left ventricular outflow tract obstruction (LVOTO) in 70% of patients during exercise (resting LVOTO in 25%). The Venturi effect from abnormal anterior mitral valve leaflet systolic anterior motion (SAM) exacerbates LVOTO and contributes to mitral regurgitation.
Arrhythmogenic Cardiomyopathy (ARVC)
ARVC results from mutations in desmosomal proteins — predominantly PKP2 (plakophilin-2, 45%), DSP (desmoplakin), DSG2 (desmoglein-2), DSC2, and JUP (plakoglobin) — disrupting cardiomyocyte cell-to-cell adhesion. Mechanical stress (exercise) triggers cardiomyocyte detachment and apoptosis, replaced by fibro-fatty infiltration predominantly in the right ventricular free wall, apex, and infundibulum (the "triangle of dysplasia").
Fibro-fatty replacement creates slow conduction zones supporting reentrant ventricular tachycardia circuits, explaining the exercise-provoked ventricular arrhythmias and sudden cardiac death risk in ARVC patients.
Restrictive Cardiomyopathy (RCM)
RCM is characterized by impaired ventricular filling with normal or reduced diastolic volumes, normal or mildly reduced systolic function, and elevated filling pressures. The pathophysiologic basis depends on etiology: infiltration (amyloid fibrils, glycolipids in Fabry disease), endomyocardial fibrosis (hypereosinophilic syndrome, radiation), or storage diseases (hemochromatosis — iron deposition impairs mitochondrial function and contractility).
Cardiac amyloidosis (transthyretin [ATTR] or light-chain [AL] amyloid) causes amyloid fibril deposition in the extracellular matrix, increasing myocardial stiffness and impairing diastolic filling. ATTR amyloid also impairs sodium channel function, contributing to conduction abnormalities.
4. Etiology and Risk Factors
Dilated Cardiomyopathy — Causes
- Genetic/familial (25–35%): TTN, LMNA, MYH7, TNNT2, SCN5A, PLN mutations
- Myocarditis: Viral (Coxsackievirus B, adenovirus, parvovirus B19, HIV), autoimmune
- Alcohol (alcoholic cardiomyopathy): Dose-dependent; >80 g/day for >5 years; partially reversible with abstinence
- Peripartum cardiomyopathy: Last month of pregnancy to 5 months postpartum; prolactin cleavage product mechanism
- Tachycardia-induced: Prolonged rapid atrial or ventricular arrhythmias; largely reversible with rate/rhythm control
- Chemotherapy-related: Anthracyclines (doxorubicin) — dose-dependent ROS-mediated cardiomyocyte injury; trastuzumab (HER2/ErbB2 blockade)
- Stress (Takotsubo): Catecholamine surge causing reversible apical ballooning
- Ischemic cardiomyopathy: Technically a secondary cardiomyopathy; most common cause of HFrEF
- Metabolic: Thyroid disorders (both hypo- and hyperthyroidism), acromegaly, pheochromocytoma
- Nutritional deficiencies: Thiamine (beriberi), selenium, carnitine
Hypertrophic Cardiomyopathy — Risk Factors
- Autosomal dominant sarcomeric gene mutations (penetrance variable)
- Family history of HCM or sudden cardiac death <50 years
- Syndromic HCM: Noonan syndrome (PTPN11, RAF1), Pompe disease (GAA), Danon disease (LAMP2), LEOPARD syndrome
Secondary Cardiomyopathy Causes
- Cardiac sarcoidosis (granulomatous infiltration, arrhythmias and conduction disease)
- Cardiac amyloidosis: Wild-type ATTR (senile systemic amyloidosis, predominates in older men), hereditary ATTR (Val30Met most common worldwide; Val122Ile in 3–4% of African Americans), AL amyloidosis (plasma cell dyscrasia)
- Hemochromatosis: HFE gene mutations, iron overload
- Fabry disease: Alpha-galactosidase A deficiency; X-linked; cardiac variant presents as HCM phenotype
- Connective tissue diseases: Systemic lupus erythematosus, systemic sclerosis
- Endomyocardial fibrosis: Tropical regions; associated with hypereosinophilia (Loeffler endocarditis)
5. Clinical Presentation
Dilated Cardiomyopathy
DCM typically presents with progressive heart failure symptoms: exertional dyspnea (NYHA Class I–IV), orthopnea, paroxysmal nocturnal dyspnea, fatigue, and reduced exercise tolerance. Physical examination reveals displaced/diffuse PMI, S3 gallop, functional mitral regurgitation murmur, elevated JVP, pulmonary crackles, and peripheral edema in advanced disease. Arrhythmias (AF, ventricular tachycardia) and conduction abnormalities are common, particularly in LMNA-related DCM.
Hypertrophic Cardiomyopathy
The triad of exertional dyspnea, chest pain, and syncope (particularly exertional syncope — a warning sign for SCD) characterizes HCM. Many patients are asymptomatic, identified by family screening or incidental ECG/echocardiography findings. Physical examination with LVOTO reveals:
- Bisferiens (double-peaked) carotid pulse
- Systolic ejection murmur at the left sternal border, increasing with Valsalva maneuver (decreased preload increases LVOTO gradient) and decreasing with squatting (increased preload) — distinguishing HCM from aortic stenosis
- Holosystolic apical murmur from SAM-related mitral regurgitation
- Sustained PMI with double apical impulse (atrial and ventricular components)
Arrhythmogenic Cardiomyopathy (ARVC)
ARVC typically presents in young adults (15–40 years) with palpitations, syncope, or sustained ventricular tachycardia (left bundle branch block morphology, suggesting RV origin). Sudden cardiac death — often exercise-triggered — can be the first manifestation. Physical examination is often normal early; signs of RV failure develop in advanced disease.
Restrictive Cardiomyopathy
RCM presents with symptoms of biventricular failure: severe exercise intolerance (dominant complaint), dyspnea, lower extremity edema, and ascites. Examination reveals elevated JVP with prominent x and y descents (Kussmaul's sign — failure of JVP to fall with inspiration), S3/S4 gallop, hepatomegaly, and peripheral edema. In cardiac amyloidosis, additional features include macroglossia (AL amyloid), periorbital purpura (AL amyloid — pathognomonic), carpal tunnel syndrome, and bilateral low-voltage ECG despite thick walls on echocardiography (classic "voltage-wall thickness discordance").
6. Diagnosis
Electrocardiography
- DCM: Left bundle branch block (LBBB), nonspecific ST-T changes, AF, ventricular ectopy; LMNA-DCM: PR prolongation, AV block, AF, ventricular arrhythmias
- HCM: LVH voltage criteria, deep Q waves (pseudo-infarction pattern in leads I, aVL, V5-V6 from septal depolarization), giant negative T waves (apical HCM — Yamaguchi pattern)
- ARVC: Epsilon wave (pathognomonic — small positive deflection after QRS in V1-V3), T-wave inversions in V1-V3, prolonged terminal activation duration >55 ms in V1-V3, LBBB-morphology VT
- Amyloidosis: Pseudo-infarction pattern (loss of R waves), low voltage (QRS amplitude <5 mm in limb leads), AF, conduction disease
Echocardiography
The primary imaging modality for initial evaluation:
- DCM: LV/biventricular dilation, reduced LVEF (<50%), reduced global longitudinal strain (GLS), functional MR/TR
- HCM: Maximal wall thickness ≥15 mm (or ≥13 mm with family history/genetic confirmation), typically asymmetric, LVOTO gradient ≥30 mmHg at rest or ≥50 mmHg with provocation, SAM of mitral valve, mitral regurgitation; apical HCM: apical obliteration; spade-shaped LV cavity on contrast ventriculography
- ARVC: RV dilation, regional RV wall motion abnormalities (akinesia/dyskinesia), RV outflow tract (RVOT) dilation, reduced RVEF; often subtle early
- Amyloidosis: Concentric LV wall thickening, "sparkling" or granular myocardial appearance, biatrial enlargement, thickened valves, pericardial effusion, RV hypertrophy, impaired GLS with relative apical sparing (apical sparing pattern — high sensitivity and specificity for cardiac amyloidosis)
Cardiac Magnetic Resonance Imaging (CMR)
CMR is the gold standard for myocardial characterization, tissue quantification, and ARVC evaluation:
- Late gadolinium enhancement (LGE): Identifies myocardial fibrosis; mid-wall or epicardial LGE in DCM; patchy LGE in HCM (predicts SCD risk); RV LGE in ARVC; subendocardial/transmural LGE in ischemic cardiomyopathy; diffuse subendocardial LGE in amyloidosis
- T1 mapping and extracellular volume fraction (ECV): Quantifies diffuse fibrosis and amyloid infiltration; ECV >40% highly specific for cardiac amyloidosis
- T2 mapping: Identifies myocardial edema and inflammation (myocarditis, acute phase)
- Fat/fibrosis quantification in ARVC: Fatty replacement of RV myocardium (T1-weighted imaging)
- ARVC Task Force Criteria (2010, revised 2020): Incorporates CMR findings (RV functional and structural criteria) alongside ECG, biopsy, arrhythmia, and family history criteria to diagnose definite, borderline, or possible ARVC
Additional Diagnostic Testing
- Genetic testing: Recommended for all patients with DCM, HCM, and ARVC; guides family screening; impacts prognosis (LMNA-DCM: high risk of SCD and conduction disease; ARVC: confirms diagnosis and risk stratifies)
- Endomyocardial biopsy (EMB): Indicated for unexplained new-onset HF with hemodynamic compromise (to rule out giant cell myocarditis), unexplained ARVC, suspected infiltrative disease (amyloidosis confirmation), or hemochromatosis; diagnostic yield variable by pathology
- Nuclear imaging (PYP scintigraphy): Technetium-99m pyrophosphate (Tc-PYP) scan — highly sensitive and specific for ATTR cardiac amyloidosis (Perugini grade 2–3 in absence of AL amyloidosis by serum/urine immunofixation and free light chains); widely used for non-invasive ATTR diagnosis
- Holter monitoring and implantable loop recorder: Arrhythmia characterization; high-risk ARVC screening
- Cardiopulmonary exercise testing (CPET): Functional assessment (peak VO₂); HCM exercise provocation for dynamic LVOTO gradient; prognosis in DCM (peak VO₂ <10 mL/kg/min — listing criteria for transplantation)
- Serum biomarkers: NT-proBNP/BNP (severity/prognosis); troponin (ongoing myocyte injury); serum/urine protein electrophoresis and free light chain ratio (AL amyloidosis); serum ATTR (transthyretin) level; ferritin/transferrin saturation (hemochromatosis); alpha-galactosidase A activity (Fabry disease)
7. Treatment
Dilated Cardiomyopathy
DCM treatment follows guideline-directed medical therapy (GDMT) for HFrEF (see Heart Failure guidelines):
- ACE inhibitors/ARBs/ARNI: Sacubitril/valsartan (Entresto) preferred over ACE inhibitor/ARB when tolerated; reduces mortality 20% (PARADIGM-HF)
- Beta-blockers: Carvedilol, metoprolol succinate, bisoprolol; mortality benefit proven in multiple RCTs
- Mineralocorticoid receptor antagonists (MRA): Spironolactone, eplerenone; eGFR >30 mL/min/1.73m², K+ <5.0 mEq/L
- SGLT2 inhibitors: Dapagliflozin, empagliflozin; 25% relative risk reduction in HF hospitalization/CV death; now class I for all HFrEF regardless of diabetes
- Cardiac resynchronization therapy (CRT): LBBB, QRS ≥150 ms, EF ≤35%, NYHA Class II–IV; reduces mortality and improves EF
- Implantable cardioverter-defibrillator (ICD): Primary prevention — EF ≤35%, NYHA II–III on GDMT for >3 months; secondary prevention after documented life-threatening ventricular arrhythmia
- Peripartum cardiomyopathy: Standard HF therapy (avoid ACE inhibitors during pregnancy; use hydralazine/nitrates); bromocriptine 2.5 mg twice daily for 4 weeks (BOARD trial) — suppresses prolactin, improves recovery; anticoagulation while EF <35% or LV thrombus
- Heart transplantation: End-stage DCM refractory to GDMT; peak VO₂ <10 mL/kg/min; 5-year survival ~75% post-transplantation
- Left ventricular assist device (LVAD): Bridge-to-transplantation or destination therapy in Stage D HF
Hypertrophic Cardiomyopathy
- Symptomatic LVOTO (resting gradient ≥30 mmHg or provokable ≥50 mmHg):
- Beta-blockers (first-line): Metoprolol succinate, atenolol, propranolol; reduce heart rate and LVOTO gradient
- Non-dihydropyridine CCBs: Verapamil (when beta-blockers not tolerated); negative inotropic and chronotropic effects
- Disopyramide: Class IA antiarrhythmic; negative inotrope; 50–300 mg twice daily (extended-release); anticholinergic side effects; used in combination with beta-blockers
- Mavacamten (cardiac myosin inhibitor): FDA-approved 2022 (EXPLORER-HCM trial); reduces LVOTO gradient and improves symptoms (NYHA class, QoL, exercise capacity); first disease-modifying therapy for HCM; REMS program required; EF monitoring mandatory (hold if EF <50%)
- Aficamten (next-generation myosin inhibitor): Shorter half-life than mavacamten; SEQUOIA-HCM trial (2024) demonstrated significant LVOTO gradient reduction
- Septal reduction therapy for drug-refractory obstructive HCM (gradient ≥50 mmHg with NYHA III–IV):
- Surgical septal myectomy (Morrow procedure): Gold standard at experienced centers (>200 procedures/lifetime); 90% symptom relief; operative mortality <1%; concomitant MV repair/replacement if needed
- Alcohol septal ablation: Percutaneous technique; alcohol injection into septal perforating arteries causes controlled infarction; suitable for elderly/high surgical risk; higher rate of LBBB and complete heart block requiring permanent pacemaker (10–20%)
- SCD prevention in HCM: ICD implantation for primary prevention in patients with ≥1 major SCD risk factor: prior cardiac arrest or sustained VT, family history of SCD, unexplained syncope, LV wall thickness ≥30 mm, hypotensive blood pressure response to exercise, non-sustained VT on Holter, or extensive LGE on CMR (>15% LV mass). The 2022 AHA/ACC HCM Guideline recommends using the HCM Risk-SCD calculator (European) or a multifactorial clinical assessment (US) to individualize ICD decisions.
- Non-obstructive HCM: Symptom management with beta-blockers or CCBs; no proven disease-modifying therapy beyond mavacamten (currently only approved for obstructive HCM)
- Atrial fibrillation in HCM: Anticoagulation for all patients with HCM and AF regardless of CHA₂DS₂-VASc score (Class I); rhythm control preferred given poor tolerance of AF in HCM
Arrhythmogenic Cardiomyopathy (ARVC)
- Restriction from competitive sports and endurance exercise: Exercise accelerates fibro-fatty progression; families should undergo cascade screening and genetic testing
- Beta-blockers: Sotalol or nadolol for arrhythmia suppression; sotalol (class III) preferred for sustained VT suppression
- ICD: Secondary prevention for documented VF/sustained VT; primary prevention criteria debated (history of unexplained syncope, extensive RV involvement, desmosomal compound heterozygosity)
- Catheter ablation: VT ablation (endocardial and epicardial) for recurrent ICD shocks or drug-refractory VT; adjunct to ICD therapy
- Heart transplantation: End-stage biventricular failure or uncontrolled ventricular arrhythmias
Cardiac Amyloidosis (ATTR)
- Tafamidis (transthyretin stabilizer): 80 mg once daily; reduces all-cause mortality by 30% and HF hospitalization by 32% in ATTR amyloidosis (ATTR-ACT trial, 2018); FDA-approved for wild-type and hereditary ATTR cardiomyopathy; only pharmacotherapy with proven cardiac survival benefit in ATTR
- Patisiran (siRNA-based TTR silencer): Reduces TTR production; APOLLO-B trial (2022) — stabilizes or improves 6-minute walk distance and QoL; FDA-approved for hATTR polyneuropathy; cardiac efficacy data accumulating
- Eplontersen/vutrisiran: Next-generation subcutaneous siRNA agents; convenient monthly dosing; phase III cardiac trials ongoing
- Acoramidis: Next-generation TTR stabilizer (ATTRibute-CM trial, 2023: 50% reduction in all-cause mortality + CV hospitalization at 30 months); FDA approved November 2024
- Diuretics: Essential for volume management; ACE inhibitors and beta-blockers often poorly tolerated due to vasodilatory and negative chronotropic effects on already impaired cardiac output
- Anticoagulation: High AF prevalence in amyloidosis; DOAC preferred but specific data limited
- AL amyloidosis: Plasma cell-directed therapy (bortezomib-based regimens, daratumumab-based regimens, autologous stem cell transplantation in eligible patients) to suppress light chain production
8. Complications
- Sudden cardiac death (SCD): Leading cause of mortality in HCM, ARVC, and LMNA-DCM; underlying mechanism is ventricular fibrillation or polymorphic VT
- Heart failure progression: End-stage DCM with refractory HFrEF; progressive symptoms in amyloidosis
- Atrial fibrillation: Highly prevalent in all cardiomyopathy subtypes; particularly poorly tolerated in HCM (loss of atrial kick in stiff, non-compliant ventricle) and amyloidosis
- Ventricular tachycardia/fibrillation: Life-threatening arrhythmias; ARVC and DCM with LGE are particularly high-risk
- Thromboembolic events: LV thrombus with systemic embolism; particularly in DCM with EF <35%; warfarin or DOAC anticoagulation indicated for LV thrombus
- Conduction disease: LMNA-DCM — progressive AV block, sick sinus syndrome; cardiac amyloidosis — AV and infra-Hisian conduction disease; often requires pacemaker implantation
- Mitral regurgitation: SAM-related in HCM (dynamic); functional MR in DCM from annular dilation and papillary muscle displacement
- Progressive RV failure: Biventricular involvement in amyloidosis, sarcoidosis, ARVC; portends severe prognosis
- Outflow tract obstruction in HCM: Acute hemodynamic compromise (especially with dehydration, vasodilators); worsens with Valsalva, standing, tachycardia
9. Prognosis
- DCM: 5-year mortality with optimal GDMT approximately 20–30%; annual SCD risk 2–4%; LMNA-DCM has worst prognosis among genetic DCMs — 50% develop life-threatening arrhythmias by age 50
- HCM: Annual mortality approximately 1–2% in contemporary cohorts with appropriate ICD implantation; SCD risk is highest in children and adolescents; "burnt-out" HCM (LV dilation, reduced EF) carries poor prognosis similar to DCM
- ARVC: Annual SCD risk 1–2.5% in symptomatic patients; ICD reduces SCD mortality; end-stage biventricular failure develops in 10–15% over 20 years
- ATTR cardiac amyloidosis: Without tafamidis, median survival ~2.5–3.5 years from diagnosis; with tafamidis, 30% survival benefit at 2.5 years; AL amyloidosis has worse cardiac prognosis (median survival 6 months in cardiac AL without treatment; response to plasma cell therapy dramatically improves outcomes)
- Peripartum cardiomyopathy: EF recovery in 50% at 6 months; subsequent pregnancy carries 30–50% recurrence risk; EF <30% at presentation associated with persistent LV dysfunction
- Takotsubo cardiomyopathy: In-hospital mortality 4–5%; EF typically fully recovers within 4–8 weeks; recurrence risk 10% over 5 years; prognosis predominantly determined by precipitating illness
10. Prevention
Primary Prevention
- Genetic counseling and cascade family screening: All first-degree relatives of patients with DCM, HCM, or ARVC; cascade screening with genetic testing and echocardiography
- Alcohol moderation: Prevents alcoholic DCM; abstinence reverses established alcoholic cardiomyopathy in 50–80%
- Cardioprotection during chemotherapy: Cardio-oncology co-management; baseline and serial cardiac surveillance; dexrazoxane for anthracycline cardioprotection; early GDMT initiation for subclinical LV dysfunction
- Sports restriction in genetic cardiomyopathies: ARVC and HCM patients should avoid competitive and high-intensity endurance sports
- ATTR screening: Annual echocardiography and Tc-PYP scan in patients over 60 with LV hypertrophy and HF symptoms; genetic testing of Val122Ile in African American patients with unexplained LVH
Secondary Prevention
- Optimized GDMT to prevent DCM progression
- ICD for SCD prevention in high-risk HCM and ARVC
- Anticoagulation for AF in all cardiomyopathy subtypes
- Wearable cardioverter-defibrillator (WCD) for new-onset DCM during initial 3–6-month GDMT optimization period (before reevaluating ICD indication)
- Exercise prescription: Moderate-intensity exercise for DCM; restriction for HCM with LVOTO and ARVC
11. Recent Research and Advances
- Mavacamten (EXPLORER-HCM, VALOR-HCM, 2022–2023): First cardiac myosin inhibitor approved for obstructive HCM; VALOR-HCM demonstrated mavacamten substantially reduced the proportion of patients meeting criteria for septal reduction therapy at 16 weeks (17.9% vs. 76.8% placebo). A paradigm shift toward medical alternatives to invasive septal reduction therapy.
- Acoramidis (ATTRibute-CM, 2023): Second transthyretin stabilizer; 50% relative reduction in composite of all-cause mortality and CV hospitalization vs. placebo; FDA-approved November 2024, expanding ATTR treatment options beyond tafamidis.
- Gene therapy for DCM and ARVC: AAV9-PLN (phospholamban inhibition) in early trials for PLN-cardiomyopathy; AAV-PKP2 for ARVC-PKP2 patients (RESCUE trial); CRISPR-based gene editing for TTN truncating variants in preclinical models.
- Cardiac RNA therapeutics: Eplontersen (subcutaneous antisense oligonucleotide) and vutrisiran (subcutaneous siRNA) target TTR mRNA; once-monthly dosing improves adherence vs. patisiran (IV every 3 weeks); HELIOS-B trial (vutrisiran in cardiac ATTR, 2024) — positive results.
- RESET HCM trial: Evaluating mavacamten vs. myectomy in obstructive HCM to determine optimal sequencing of medical vs. invasive therapy.
- ARVC genetics and exercise: Definitive demonstration that competitive athletic activity accelerates ARVC disease expression and penetrance; 2023 ACC/AHA guidelines recommend shared decision-making for recreational exercise but advise against competitive sports for ARVC gene carriers even before phenotypic expression.
- Artificial intelligence in cardiomyopathy: AI-ECG algorithms achieve ~85% sensitivity for detecting LVEF <35% (DCM screening); AI-echocardiography tools automate GLS and ECV quantification; AI pathology for automated HCM biopsy interpretation.
- Sotatercept: Activin receptor type II-A ligand trap; phase II data in cardiac amyloidosis; primarily developed for pulmonary arterial hypertension but cardiac remodeling effects under investigation.
Research Papers
The following PubMed topic searches return current peer-reviewed literature relevant to this condition. Each link opens a live PubMed query.
- Dilated cardiomyopathy
- Hypertrophic cardiomyopathy
- Restrictive cardiomyopathy
- Arrhythmogenic right ventricular cardiomyopathy
- Genetic cardiomyopathy
- Cardiomyopathy guidelines
- Cardiomyopathy cardiac MRI
- Peripartum cardiomyopathy
- Ischemic cardiomyopathy
- Takotsubo cardiomyopathy
- Cardiomyopathy mortality
- Cardiomyopathy treatment
Connections
- Cardiology
- How Heart Failure Develops — interactive animation
- Arrhythmia
- Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
- Heart Failure
- Myocarditis
- Sudden Cardiac Death
- Chagas Disease
- Atrial Fibrillation
- Hypertension
- Valvular Heart Disease
- Hemochromatosis
- Chest Pain
- Shortness of Breath
- Edema
- Fatigue
- Magnesium
- Omega-3 Fatty Acids
- Thyroid Disorders
- Diabetes
- Iron
- Selenium
- Heart Palpitations