Cushing's Syndrome
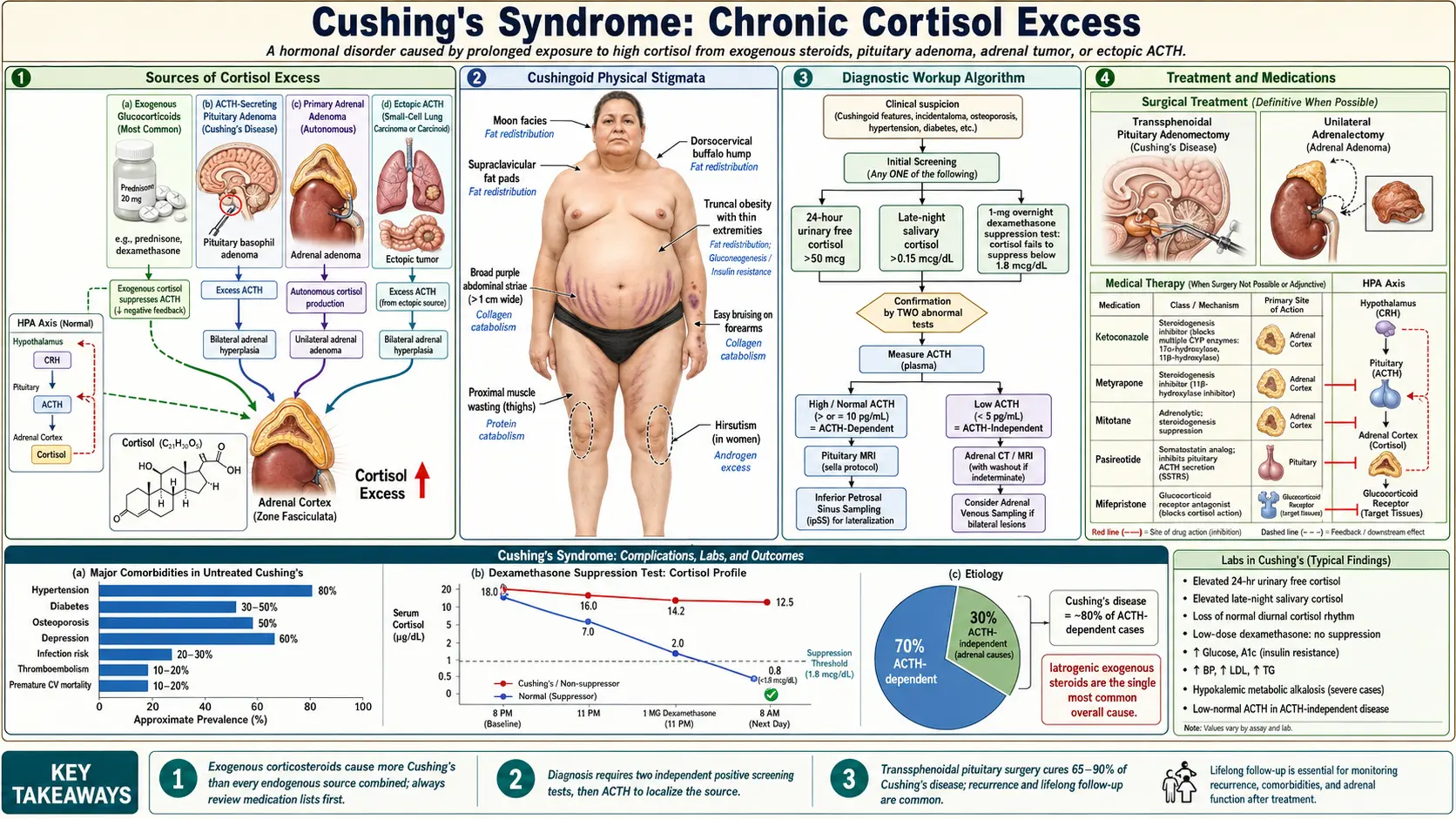
Cushing's syndrome is what happens when your body is exposed to too much cortisol for too long — whether that cortisol comes from a tumor inside you, or from a steroid medication you are taking for another condition. The result is a distinctive cluster of changes: weight that piles up in the middle of the body while the arms and legs stay thin, skin so fragile it bruises and tears from almost nothing, muscles that go weak, blood pressure that climbs, and a mood that can swing from anxiety to deep depression. What makes Cushing's particularly tricky is how often it is missed. The symptoms appear gradually and overlap with much more common problems like obesity, diabetes, and depression, so the average person waits years before getting the right diagnosis. The causes vary enormously — a tiny benign tumor on the pituitary gland, a tumor on one adrenal gland, a cancer in the lung secreting hormones, or simply long-term steroid therapy for asthma or arthritis — and each cause calls for a different treatment. The good news is that once the source of the excess cortisol is found and removed or blocked, most people recover substantially, though it can take a year or more for the body to fully recalibrate.
Interactive Visualization The Stress Response — fire a stressor and watch cortisol rise Adrenaline spikes in a second, cortisol follows in minutes — then compare acute stress with chronic, and watch the feedback loop blunt. Launch → Interactive Visualization Your Daily Cortisol Rhythm — ride the 24-hour curve Ride the 24-hour cortisol curve from its midnight low to the morning peak that wakes you up — then work a night shift or read the test at the wrong hour and see why timing is everything. Launch →
Table of Contents
- What Cushing's Syndrome Is
- ACTH-Dependent Causes
- ACTH-Independent Causes
- Classic Clinical Features
- Screening Tests
- Confirmatory Testing and Localization
- Treatment Options
- Medical Therapy for Inoperable Cases
- Research Papers
- Connections
- Featured Videos
What Cushing's Syndrome Is
Cortisol is the body's primary stress hormone. In short bursts it is lifesaving — it raises blood sugar for quick energy, sharpens focus, curbs inflammation, and mobilizes the immune system. The problem arises when cortisol is elevated not for minutes or hours but for months or years. Prolonged high cortisol rewires metabolism, redistributes fat in a characteristic pattern, breaks down muscle and bone, raises blood pressure, impairs wound healing, suppresses the immune system in harmful ways, and disrupts mood and cognition.
The term Cushing's syndrome refers to the full clinical picture of chronic glucocorticoid excess, regardless of cause. When the cause is specifically a pituitary tumor overdriving cortisol production, it is called Cushing's disease — a subset of the broader syndrome named after the neurosurgeon Harvey Cushing, who first described it in 1932.
The most important first distinction is between exogenous and endogenous Cushing's syndrome:
- Exogenous (iatrogenic) — caused by glucocorticoid medications such as prednisone, dexamethasone, or inhaled and topical steroids used at high doses or for prolonged periods. This is by far the most common cause overall and is often overlooked because the focus stays on the condition being treated rather than the side effects of treatment.
- Endogenous — the body itself is manufacturing too much cortisol. The source may be the pituitary gland, an adrenal gland, or a non-pituitary tumor elsewhere in the body secreting hormones that drive cortisol production. Endogenous Cushing's is rare, affecting roughly 10–15 people per million per year, but is serious and requires careful workup to identify and remove the source.
Within endogenous Cushing's, cases are classified by whether the excess cortisol is being driven by ACTH (adrenocorticotropic hormone) — the pituitary signal that tells the adrenal glands to make cortisol — or whether the adrenal glands are acting autonomously without any ACTH stimulus.
ACTH-Dependent Causes
About 80% of endogenous Cushing's syndrome is ACTH-dependent — meaning a tumor somewhere is secreting ACTH, which then drives the adrenal glands to overproduce cortisol. There are two main sources.
Cushing's Disease (Pituitary Corticotroph Adenoma)
This is the single most common cause of endogenous Cushing's syndrome, accounting for roughly 70% of all endogenous cases. A benign adenoma arises from the ACTH-secreting (corticotroph) cells of the anterior pituitary gland and secretes ACTH in excess, chronically stimulating both adrenal glands to enlarge and overproduce cortisol. These tumors are typically small — most are microadenomas under 10 mm — which is one reason pituitary MRI misses them in nearly half of proven cases. Women in their 30s and 40s are affected far more often than men, at a ratio of roughly 3:1.
A key feature of pituitary Cushing's disease is that the tumor retains some sensitivity to feedback regulation. It will suppress cortisol production partially in response to very high doses of dexamethasone (the high-dose dexamethasone suppression test), which is a useful diagnostic clue to distinguish it from ectopic ACTH secretion.
Ectopic ACTH Syndrome
In roughly 10–15% of endogenous Cushing's cases, ACTH is not coming from the pituitary at all but from a non-pituitary tumor elsewhere in the body. Common sources include:
- Small cell lung cancer — the most aggressive cause; cortisol rises so rapidly that classic Cushingoid fat redistribution may not have time to develop, and the dominant picture is severe hypokalemic alkalosis, profound muscle weakness, hyperpigmentation, and weight loss.
- Bronchial carcinoid tumors — slower-growing; these often produce a full Cushingoid appearance indistinguishable from Cushing's disease clinically, making localization challenging.
- Islet cell tumors of the pancreas (pancreatic neuroendocrine tumors).
- Pheochromocytoma — rare but documented; ACTH secretion from an adrenal medullary tumor.
- Thymic carcinoids, medullary thyroid carcinoma, and other neuroendocrine tumors.
Ectopic ACTH tumors do not suppress on high-dose dexamethasone testing, and inferior petrosal sinus sampling (IPSS) shows a peripheral rather than central ACTH gradient — two features that separate them from pituitary disease. Finding the ectopic source can require whole-body CT, octreotide scintigraphy, or PET scanning, and sometimes it remains occult for years despite diligent searching.
ACTH-Independent Causes
About 20% of endogenous Cushing's syndrome is ACTH-independent — the adrenal glands are making excess cortisol on their own without any signal from the pituitary. In these cases, measured plasma ACTH will be suppressed to very low or undetectable levels because the high cortisol is feeding back to shut down the hypothalamic-pituitary axis.
Unilateral Adrenal Adenoma
This is the most common ACTH-independent cause, typically a well-encapsulated, benign, cortisol-secreting adrenal adenoma. Because the contralateral adrenal gland is suppressed by the chronically elevated cortisol, bilateral adrenalectomy is not required — removing the affected gland usually cures the syndrome, though the suppressed normal adrenal may take months to recover, requiring temporary glucocorticoid replacement post-surgery.
Adrenal Carcinoma
Adrenal cortical carcinoma is rare but aggressive, with a poor prognosis. It tends to present with a larger adrenal mass (often >4 cm) and may co-secrete androgens alongside cortisol, producing virilization (deepened voice, clitoromegaly, acne, hirsutism) — a feature that should always raise suspicion for malignancy rather than a benign adenoma. Mitotane is the key adrenolytic agent used for this tumor.
Bilateral Adrenal Hyperplasia (Rare Genetic Forms)
Two rare conditions cause autonomous bilateral adrenal cortisol excess:
- Primary Pigmented Nodular Adrenocortical Disease (PPNAD) — often occurs in young patients, associated with Carney Complex (a genetic syndrome with cardiac myxomas, skin pigmentation, and other tumors). A paradoxical increase in cortisol after dexamethasone administration (Liddle's test) is characteristic.
- ACTH-Independent Macronodular Adrenal Hyperplasia (AIMAH / PBMAH) — the adrenal glands develop multiple large nodules that secrete cortisol in response to aberrant receptors (for GIP, vasopressin, catecholamines, LH, etc.) rather than ACTH. Often presents as incidentally discovered bilateral adrenal masses on imaging done for another reason.
Classic Clinical Features
The clinical picture of Cushing's syndrome spans almost every organ system, because glucocorticoid receptors are present throughout the body. The following features, particularly when several occur together, are the clues that should prompt cortisol testing.
Fat Redistribution — The Characteristic Appearance
- Central/truncal obesity — fat accumulates in the abdomen, chest, and face while the limbs remain relatively thin, giving a lemon-on-toothpicks silhouette.
- Moon facies — rounding and fullness of the face from fat deposition in the cheeks and temples.
- Buffalo hump (cervicodorsal fat pad) — a prominent fat deposit at the base of the neck and upper back.
- Supraclavicular fat pads — fat filling the hollows above the collarbones; this sign is highly specific for Cushing's when present.
Skin and Soft Tissue Signs
- Violaceous (purple) striae — stretch marks wider than 1 cm, typically on the abdomen, thighs, breasts, and axillae; the purple-red color reflects dermal atrophy and visible underlying blood vessels. Ordinary stretch marks are thinner and pinker.
- Easy bruising — the skin becomes thin and fragile from collagen loss; even minor trauma leaves large bruises.
- Impaired wound healing — cuts and surgical incisions heal slowly and poorly.
- Acne and hirsutism — driven by androgen co-secretion or direct glucocorticoid effects.
- Skin hyperpigmentation — seen in ACTH-dependent forms (especially ectopic) because ACTH and melanocyte-stimulating hormone (MSH) share the same precursor molecule (POMC).
Muscle and Bone
- Proximal myopathy — weakness of the hip flexors and shoulder girdle muscles, so the person has difficulty rising from a low chair or climbing stairs without using the arms. This is one of the most diagnostically specific features of Cushing's.
- Osteoporosis and fractures — cortisol inhibits osteoblasts and promotes bone resorption; vertebral compression fractures can be the presenting problem, sometimes in relatively young patients.
Metabolic and Cardiovascular
- Hypertension — cortisol has mineralocorticoid activity and sensitizes blood vessels to catecholamines; hypertension is present in the large majority of patients and contributes significantly to cardiovascular risk.
- Glucose intolerance and type 2 diabetes — cortisol drives hepatic gluconeogenesis and induces insulin resistance; frank diabetes occurs in roughly 50% of patients.
- Hypokalemic alkalosis — particularly pronounced in ectopic ACTH syndrome, where cortisol levels are extremely high and overwhelm the enzyme (11β-HSD2) that normally protects mineralocorticoid receptors from cortisol.
- Dyslipidemia and hypercoagulability — increased risk of deep vein thrombosis and pulmonary embolism.
Reproductive and Endocrine
- Menstrual irregularity and amenorrhea in women; reduced libido and erectile dysfunction in men — cortisol suppresses GnRH and the hypothalamic-pituitary-gonadal axis.
- Hirsutism, acne, clitoromegaly, and deepened voice when significant androgen co-secretion is present, which points toward adrenal carcinoma.
Neuropsychiatric Features
- Depression, anxiety, emotional lability — among the earliest and most disabling symptoms; cortisol alters monoamine neurotransmission and hippocampal structure.
- Cognitive impairment — difficulties with memory and concentration; hippocampal volume loss is measurable on MRI and partially reversible after cure.
- Psychosis — less common but can be the presenting feature, especially with very high cortisol levels.
Immune Suppression
- Increased susceptibility to opportunistic infections — Pneumocystis jirovecii pneumonia, oral candidiasis, and unusual bacterial infections are red flags in an undiagnosed patient.
- Reactivation of latent tuberculosis.
Screening Tests
Screening for Cushing's syndrome is recommended when a patient has multiple features of cortisol excess, particularly the more discriminating ones (proximal myopathy, violaceous striae, easy bruising, or unexplained osteoporosis in a young person). A positive result on any one of the following first-line tests should be followed by a second confirmatory test. Because no single test is perfect, the Endocrine Society recommends using at least two tests to confirm the diagnosis.
24-Hour Urine Free Cortisol (UFC)
Measures the total cortisol excreted in the urine over a full day, reflecting integrated cortisol secretion. A result more than three times the upper limit of normal is highly specific for Cushing's syndrome. The test requires a complete 24-hour urine collection (incomplete collections are a common source of false-negative results) and should be repeated at least twice because cortisol secretion in Cushing's syndrome can be episodic. This test is less reliable in significant renal impairment (GFR <60 mL/min) because cortisol excretion is reduced.
Late-Night Salivary Cortisol
Cortisol normally follows a strong circadian rhythm — high in the morning and lowest around midnight. In Cushing's syndrome, this nighttime nadir is lost. Salivary cortisol collected at 11 pm on two separate occasions is a convenient and sensitive screening test that can be done at home. The saliva sample is stable at room temperature and mailed to the lab. Two elevated results on different nights significantly increase diagnostic confidence.
1 mg Overnight Dexamethasone Suppression Test (Low-Dose DST)
The patient takes 1 mg of dexamethasone at 11 pm; a blood cortisol is drawn the next morning at 8 am. In a normal person, the dexamethasone mimics the feedback signal that shuts off cortisol production, and the morning cortisol suppresses to below 1.8 µg/dL. In Cushing's syndrome — where the tumor is at least partially resistant to this feedback — the morning cortisol remains above 1.8 µg/dL. This test is sensitive (catches most cases) but has a modest false-positive rate in patients who are obese, depressed, alcoholic, or taking medications that accelerate dexamethasone metabolism (rifampicin, anticonvulsants).
All three tests can be falsely abnormal in pseudo-Cushing's states — conditions that activate the HPA axis without a tumor, particularly severe depression, alcoholism, poorly controlled diabetes, and chronic stress. Distinguishing pseudo-Cushing's from true Cushing's can require CRH stimulation testing or a period of watchful waiting.
Confirmatory Testing and Localization
Once biochemical Cushing's syndrome is confirmed, the next question is: where is the source? This requires a combination of dynamic hormonal tests and imaging.
Measuring Plasma ACTH
This single measurement divides cases immediately:
- ACTH suppressed (<5–10 pg/mL) — adrenal source; proceed to adrenal CT.
- ACTH normal or elevated — pituitary or ectopic source; proceed to pituitary MRI and high-dose DST.
High-Dose Dexamethasone Suppression Test (8 mg Overnight)
The patient takes 8 mg of dexamethasone at 11 pm; a morning cortisol is drawn the next day. In Cushing's disease (pituitary adenoma), the tumor retains some feedback sensitivity, and cortisol will usually suppress by at least 50% from baseline. In ectopic ACTH secretion, the tumor is completely autonomous and cortisol does not suppress. This test has significant overlap between the two groups and is most useful as part of a panel rather than in isolation.
CRH Stimulation Test
Intravenous CRH (corticotropin-releasing hormone) stimulates ACTH secretion from the normal pituitary and from pituitary adenomas (Cushing's disease), but does not stimulate most ectopic ACTH-secreting tumors. An exaggerated ACTH and cortisol response to CRH strongly supports a pituitary source.
Inferior Petrosal Sinus Sampling (IPSS)
This is the gold standard for distinguishing pituitary Cushing's disease from ectopic ACTH syndrome when imaging and dynamic tests give ambiguous results. Catheters are placed bilaterally into the inferior petrosal sinuses — the venous drainage of the pituitary gland — and blood is sampled simultaneously from both petrosal sinuses and a peripheral vein, before and after CRH injection.
- A central-to-peripheral ACTH ratio >2 at baseline or >3 after CRH confirms a pituitary source with high accuracy (>95%).
- A ratio below these thresholds suggests ectopic ACTH secretion.
- A right-to-left gradient between the two petrosal sinuses can lateralize the pituitary adenoma, helping the surgeon plan the approach.
Imaging
- Pituitary MRI with gadolinium — the first-line imaging test for suspected Cushing's disease. However, it fails to visualize the adenoma in approximately 40–50% of proven cases because the tumors are so small. A negative MRI does not exclude Cushing's disease.
- CT of the chest and abdomen/pelvis — used to search for adrenal tumors and ectopic ACTH-secreting tumors (lung, thymus, pancreas).
- Adrenal CT or MRI — when ACTH is suppressed; characterizes adenoma vs carcinoma (size, lipid content, washout characteristics on CT).
- Octreotide scintigraphy or Ga-68 DOTATATE PET — for occult ectopic ACTH tumors, particularly carcinoids, which express somatostatin receptors.
Treatment Options
Treatment is directed at removing or ablating the source of excess cortisol. The approach differs substantially by cause.
Transsphenoidal Surgery (TSS) for Cushing's Disease
Surgery to remove the pituitary adenoma through the nose and sphenoid sinus is the first-line treatment for Cushing's disease. In experienced pituitary centers, remission rates of 70–80% are achieved for microadenomas. Recurrence occurs in roughly 15–25% of patients over 10 years. After successful TSS, the suppressed normal corticotroph cells need months to recover, so patients must take hydrocortisone replacement therapy and be monitored carefully for adrenal insufficiency during this recovery period.
Adrenalectomy for Adrenal Causes
- Unilateral adrenalectomy — curative for unilateral adenoma or carcinoma. Laparoscopic approach is standard for benign lesions; open surgery is preferred for carcinoma to achieve complete oncological margins.
- Bilateral adrenalectomy — used when the primary ectopic ACTH source cannot be found or resected, and when other options have failed. Immediately and permanently cures hypercortisolism but creates lifelong adrenal insufficiency requiring permanent glucocorticoid and mineralocorticoid replacement. A critical risk is Nelson's syndrome — after both adrenal glands are removed, the feedback restraint on the residual pituitary adenoma is lost, the tumor can grow aggressively, and ACTH levels rise dramatically, causing severe hyperpigmentation. Prophylactic pituitary radiation is often considered before bilateral adrenalectomy to reduce this risk.
Resection of Ectopic ACTH Source
When the ectopic tumor is localized (bronchial carcinoid, thymic carcinoid, islet cell tumor), surgical resection is curative. When the source is small cell lung cancer or an occult tumor that cannot be found, medical therapy or bilateral adrenalectomy is pursued instead.
Pituitary Radiation
Stereotactic radiosurgery (Gamma Knife, CyberKnife) achieves remission in approximately 50–60% of patients with persistent or recurrent Cushing's disease after TSS, though it can take 1–3 years to take full effect. Conventional fractionated radiotherapy is less preferred due to higher rates of hypopituitarism over time. Radiation is generally used as a second-line measure after failed surgery.
Medical Therapy for Inoperable Cases
When surgery is not immediately possible, has failed, or when a patient needs rapid cortisol lowering before surgery, a range of medications that block cortisol production or action are available.
Steroidogenesis Inhibitors
- Ketoconazole — the most widely used oral agent globally; inhibits multiple cytochrome P450 enzymes in the steroidogenesis pathway, reducing cortisol synthesis. Hepatotoxicity (including rare fulminant hepatic failure) is the main concern; liver enzymes must be monitored closely. Effective in most patients at controlling cortisol.
- Metyrapone — blocks the final step of cortisol synthesis (11β-hydroxylase). Works rapidly and is useful for pre-surgical cortisol control. Can worsen hirsutism and acne by causing androgen precursor accumulation. Hypertension may worsen due to 11-deoxycorticosterone (a mineralocorticoid precursor) buildup.
- Osilodrostat — a newer, potent 11β-hydroxylase inhibitor approved specifically for Cushing's disease; faster and more potent than metyrapone with a somewhat more tolerable profile.
- Mitotane — an adrenolytic agent (derived from the pesticide DDD) that directly destroys adrenocortical cells; used primarily for adrenal carcinoma. Slow onset, significant toxicity (gastrointestinal, neurological), and causes permanent adrenal insufficiency at high doses. Requires monitoring of plasma mitotane levels.
- Etomidate — an intravenous anesthetic that also potently inhibits steroidogenesis; used in the hospital for acute, severe hypercortisolism (Cushing's crisis) when rapid cortisol control is life-saving and oral agents cannot be used.
Pituitary-Directed Agents
- Pasireotide (Signifor) — a multireceptor-targeted somatostatin analog that binds the sst5 receptor preferentially expressed on corticotroph adenomas, suppressing ACTH secretion. Approved for Cushing's disease when surgery has failed or is not an option. In the pivotal trial, urinary free cortisol normalized in approximately 15–25% of patients. The major side effect is hyperglycemia (occurs in >70% of patients) due to simultaneous suppression of insulin and glucagon secretion; glucose-lowering therapy is frequently required.
- Cabergoline — a dopamine agonist that suppresses ACTH in some corticotroph adenomas; used off-label as adjunctive therapy with variable efficacy.
Glucocorticoid Receptor Antagonist
- Mifepristone (Korlym) — blocks the glucocorticoid receptor peripherally, relieving the metabolic effects of excess cortisol (glucose intolerance, diabetes). It does not lower measured cortisol levels; in fact, cortisol levels rise further due to loss of feedback inhibition. Mifepristone is approved specifically for Cushing's syndrome with glucose intolerance or diabetes in patients who are not surgical candidates. It cannot be used to monitor biochemical response in the usual way. Also blocks progesterone receptors, causing endometrial thickening and potential hypokalemia.
Long-Term Monitoring After Cure
Even after successful surgery, patients require long-term follow-up. Recurrence must be watched for with periodic cortisol testing. The metabolic complications — hypertension, diabetes, dyslipidemia, osteoporosis — may persist for years and require targeted management. Bone density should be measured and treated proactively. Cardiovascular risk remains elevated for some time even after cortisol normalization. Neuropsychiatric symptoms often lag well behind biochemical cure and may need specific support.
Research Papers
- Lacroix A, Feelders RA, Stratakis CA, Nieman LK (2015). Cushing's syndrome. Lancet, 386(9996):913–927. PMID 26004339. doi:10.1016/S0140-6736(15)00546-4
- Nieman LK, et al (2008). The Diagnosis of Cushing's Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 93(5):1526–1540. PMID 18334580. doi:10.1210/jc.2008-1337
- Nieman LK, et al (2015). Treatment of Cushing's Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 100(8):2807–2831. PMID 26222757. doi:10.1210/jc.2015-1605
- Fleseriu M, et al (2021). Consensus on diagnosis and management of Cushing's disease. Lancet Diabetes Endocrinol, 9(12):847–875. PMID 34687601. doi:10.1016/S2213-8587(21)00235-7
- Feelders RA, Pulgar SJ, Kempel A, Pereira AM (2012). The burden of Cushing's disease: clinical and health-related quality of life aspects. Eur J Endocrinol, 167(3):311–326. PMID 22728342. doi:10.1530/EJE-12-0572
- Tsigos C, Chrousos GP (2002). Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res, 53(4):865–871. PMID 12377295. doi:10.1016/S0022-3999(02)00429-4
- Biller BM, et al (2008). Treatment of adrenocorticotropin-dependent Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab, 93(7):2454–2462. PMID 18413426. doi:10.1210/jc.2007-2734
- Arnaldi G, et al (2003). Diagnosis and complications of Cushing's syndrome: a consensus update. J Clin Endocrinol Metab, 88(12):5593–5602. PMID 14671138. doi:10.1210/jc.2003-030871
- Melmed S (2011). Pathogenesis of pituitary tumors. Nat Rev Endocrinol, 7(5):257–266. PMID 21423242. doi:10.1038/nrendo.2011.16
- Newell-Price J, Trainer P, Besser M, Grossman A (1998). The diagnosis and differential diagnosis of Cushing's syndrome and pseudo-Cushing's states. Endocr Rev, 19(5):647–672. PMID 9793762. doi:10.1210/er.19.5.647
- Colao A, et al; Pasireotide B2305 Study Group (2012). A 12-month phase 3 study of pasireotide in Cushing's disease. N Engl J Med, 366(10):914–924. PMID 22397646. doi:10.1056/NEJMoa1108536
- Pivonello R, et al (2014). The medical treatment of Cushing's disease: effectiveness of chronic pasireotide therapy. J Clin Endocrinol Metab, 99(5):1524–1534. PMID 24423355. doi:10.1210/jc.2013-2829
Connections
- Endocrinology
- Your Daily Cortisol Rhythm — interactive animation
- The Stress Response & Cortisol — interactive animation
- Adrenal Insufficiency
- Pheochromocytoma
- Primary Hyperaldosteronism
- Hypothyroidism
- Hypertension
- Type 2 Diabetes