Phosphorus for Energy Production
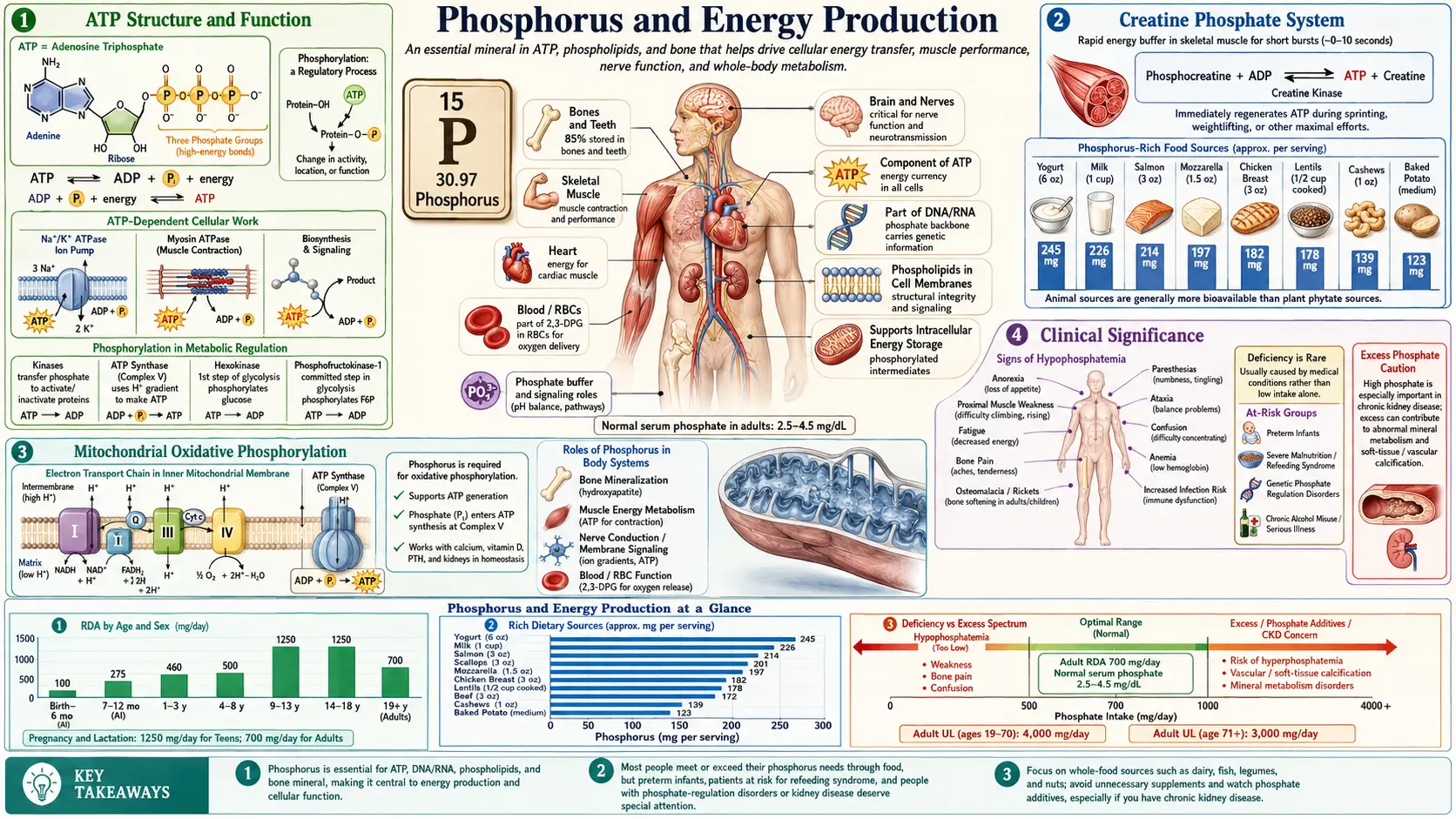
Phosphorus is the chemical core of the universal energy currency of life. Every cell on Earth uses adenosine triphosphate (ATP) — an adenosine molecule with three phosphate groups linked by high-energy phosphoanhydride bonds — to power motion, transport, biosynthesis, and signal transduction. An average adult human turns over roughly 40 to 70 kilograms of ATP per day, recycling each molecule of ATP between ADP and ATP thousands of times. Without a steady supply of phosphate, no cell could regenerate ATP, the proton-driven motor of ATP synthase would stall, and life would cease within minutes. This deep-dive walks through each layer of phosphorus-dependent energy biochemistry — ATP structure, the phosphocreatine shuttle, glycolytic intermediates, mitochondrial oxidative phosphorylation, and the clinical syndromes that result when phosphate runs out.
Table of Contents
- ATP Structure and the High-Energy Phosphoanhydride Bond
- Phosphorylation Reactions and Substrate-Level ATP
- Creatine Phosphate — the Cell's Fastest Energy Reserve
- Glycolysis and Phosphorylated Intermediates
- Mitochondrial Oxidative Phosphorylation
- Phosphorus in Coenzymes and Metabolic Pathways
- Hypophosphatemia — When ATP Synthesis Fails
- Refeeding Syndrome and the Phosphate Shift
- Clinical Applications and Repletion Strategies
- Key Research Papers
- Connections
- Featured Videos
ATP Structure and the High-Energy Phosphoanhydride Bond
Adenosine triphosphate (ATP) is a single small molecule built from three pieces: an adenine nitrogenous base, a ribose sugar, and a triphosphate tail of three sequential phosphate groups linked by phosphoanhydride bonds. The terminal (gamma) phosphate is the workhorse. Hydrolysis of the bond between the beta and gamma phosphates releases approximately 7.3 kcal/mol of free energy under standard conditions and considerably more under typical cellular conditions, where ATP is held far above its equilibrium concentration relative to ADP and inorganic phosphate (Pi).
Why is the bond so energy-rich? Three factors stack:
- Charge repulsion — at physiological pH, each phosphate group carries a net negative charge, so the three clustered phosphates of ATP repel one another. Cleaving the terminal phosphate relieves this electrostatic strain.
- Resonance stabilization — both the released inorganic phosphate (Pi) and the remaining ADP can distribute their negative charges across multiple oxygen atoms more effectively than the intact ATP can, so the products are more thermodynamically stable.
- Hydration — the separated products are better solvated by water than the intact triphosphate, releasing additional free energy as water molecules organize around the new charged surfaces.
The combination makes ATP hydrolysis exergonic enough to drive nearly any reasonable biological reaction by direct chemical coupling. The cell uses ATP in three principal ways: (1) phosphoryl transfer, where the gamma phosphate is transferred to another molecule, energizing it for the next step (this is the mechanism of nearly every kinase); (2) conformational coupling, where ATP binding and hydrolysis drive shape changes in a protein machine such as a motor protein or ion pump; and (3) adenylylation, where the AMP portion is transferred while pyrophosphate (PPi) is released and subsequently hydrolyzed, driving normally unfavorable biosynthetic reactions like the activation of amino acids onto tRNA.
GTP, UTP, and CTP are the other major nucleoside triphosphates, all phosphorus-containing, all chemically equivalent in their phosphoanhydride energetics. The cell uses different triphosphates for different processes mostly as a labeling system — GTP for protein synthesis and G-protein signaling, UTP for glycogen and other sugar metabolism, CTP for phospholipid biosynthesis — but the underlying energy is the same.
Phosphorylation Reactions and Substrate-Level ATP
Protein phosphorylation is the most common post-translational modification in eukaryotic biology. The human genome encodes more than 500 protein kinases — collectively called the "kinome" — and an estimated 30 percent or more of all human proteins are phosphorylated at any moment. Each kinase transfers the gamma phosphate from ATP to a specific serine, threonine, or tyrosine residue on a target protein, changing the protein's charge, shape, activity, localization, or binding partners.
Major phosphorylation-based signaling cascades include:
- MAP kinase pathway (RAS → RAF → MEK → ERK) — the central growth-factor signaling backbone in eukaryotic cells; dysregulation is a hallmark of cancer
- PI3K / Akt / mTOR pathway — controls cell survival, glucose uptake, and protein synthesis; deeply intertwined with insulin signaling
- JAK / STAT pathway — relays signals from cytokine receptors directly to nuclear transcription factors
- Cyclic-nucleotide pathways (PKA via cAMP, PKG via cGMP) — the classic Sutherland second-messenger system through which epinephrine, glucagon, and many other hormones act
Beyond regulatory phosphorylation, the cell uses substrate-level phosphorylation to make ATP directly. In substrate-level phosphorylation, a phosphorylated substrate transfers its phosphate to ADP, producing ATP without involvement of the electron transport chain or proton gradient. The two best-known examples are the phosphoglycerate kinase step of glycolysis (transferring the high-energy phosphate from 1,3-bisphosphoglycerate to ADP) and the succinyl-CoA synthetase step of the citric acid cycle. Substrate-level phosphorylation is quantitatively modest compared with oxidative phosphorylation but is critical for cells without functioning mitochondria (mature red blood cells), under anaerobic conditions, or for the first second of explosive exertion.
Creatine Phosphate — the Cell's Fastest Energy Reserve
The phosphocreatine system is the most rapid mechanism in biology for regenerating ATP. Creatine, a small amino-acid derivative concentrated in skeletal muscle, cardiac muscle, and brain, is phosphorylated by creatine kinase to phosphocreatine. The reaction is freely reversible:
Phosphocreatine + ADP ↔ Creatine + ATP
When ATP is suddenly consumed (the first contractions of a sprint, the first beats of a stress-induced heart-rate spike), the creatine kinase equilibrium shifts hard toward ATP production, donating the high-energy phosphate from phosphocreatine to ADP and buffering the ATP concentration before glycolysis or oxidative phosphorylation can ramp up. The reaction is essentially instantaneous and can support full-power output for roughly 5 to 10 seconds before the phosphocreatine pool is exhausted.
Beyond rapid ATP buffering, the phosphocreatine system serves as a spatial energy shuttle. Mitochondrial creatine kinase, anchored to the outer mitochondrial membrane, accepts the phosphate from newly synthesized ATP and loads it onto creatine. The resulting phosphocreatine diffuses faster through the crowded cytoplasm than ATP would and delivers the phosphate to cytoplasmic creatine kinase isoforms located adjacent to ATPases (myosin ATPase at sarcomeres, calcium-ATPase at the sarcoplasmic reticulum). This spatial coupling matters most in cells with extreme energy demand at sites distant from the mitochondrial matrix.
The clinical relevance: serum creatine kinase is one of the most important biomarkers in medicine. Elevated CK-MM signals skeletal muscle injury (rhabdomyolysis, statin myopathy, severe exercise). Elevated CK-MB signaled myocardial infarction for decades, until troponin-I and troponin-T replaced it as the preferred cardiac biomarker. Elevated CK-BB occasionally signals brain injury or some neoplasms. For more on the supplemental use of creatine to expand the phosphocreatine pool, see our Creatine page.
Glycolysis and Phosphorylated Intermediates
Every one of the ten steps of glycolysis involves a phosphorylated intermediate. The pathway is, in essence, a ten-step molecular reshuffle of phosphate groups that breaks one molecule of glucose into two molecules of pyruvate while yielding a net of two molecules of ATP (four made, two invested) and two molecules of NADH.
- Glucose → glucose-6-phosphate — hexokinase (or glucokinase in liver and pancreatic beta cells) transfers a phosphate from ATP. This irreversibly traps glucose inside the cell.
- Glucose-6-phosphate → fructose-6-phosphate — phosphoglucose isomerase rearranges the sugar.
- Fructose-6-phosphate → fructose-1,6-bisphosphate — phosphofructokinase-1 (PFK-1), the rate-limiting enzyme of glycolysis, invests a second ATP. This commits the carbon skeleton irreversibly to the pathway.
- Fructose-1,6-bisphosphate → 2 × glyceraldehyde-3-phosphate — aldolase cleaves the doubly phosphorylated six-carbon sugar into two three-carbon phosphorylated sugars.
- Glyceraldehyde-3-phosphate → 1,3-bisphosphoglycerate — glyceraldehyde-3-phosphate dehydrogenase (GAPDH) attaches a second phosphate group using inorganic phosphate (Pi) and produces NADH. This is the only step in glycolysis where free inorganic phosphate enters the pathway — making phosphate depletion a direct rate-limit on energy production.
- 1,3-bisphosphoglycerate → 3-phosphoglycerate — phosphoglycerate kinase transfers the high-energy acyl-phosphate to ADP, producing the first ATP of glycolysis (substrate-level phosphorylation).
- 3-phosphoglycerate → 2-phosphoglycerate — phosphoglycerate mutase shifts the phosphate from carbon 3 to carbon 2.
- 2-phosphoglycerate → phosphoenolpyruvate (PEP) — enolase removes water and creates a phosphate group with the highest phosphoryl transfer potential of any common biological molecule.
- PEP → pyruvate — pyruvate kinase transfers the phosphate to ADP, producing the second ATP of glycolysis (substrate-level phosphorylation).
The branch point at glucose-6-phosphate feeds the pentose phosphate pathway, which generates NADPH for reductive biosynthesis (and antioxidant defense) and ribose-5-phosphate for nucleotide synthesis. Both products contain phosphate — nothing in central carbon metabolism escapes the phosphate group.
The clinical takeaway: in severe hypophosphatemia, the GAPDH step (which requires free inorganic phosphate) becomes rate-limiting. Glycolytic ATP production grinds down, and tissues that depend on glycolysis (especially red blood cells, which have no mitochondria at all) suffer first.
Mitochondrial Oxidative Phosphorylation
Oxidative phosphorylation is the highest-yielding ATP production process in human biology, generating approximately 30 to 32 molecules of ATP per molecule of glucose oxidized (compared with the net 2 from glycolysis alone). The mechanism couples electron transport through the four respiratory complexes of the inner mitochondrial membrane with the active pumping of protons (H+) out of the mitochondrial matrix and into the intermembrane space. The resulting electrochemical proton gradient (the "proton motive force") drives ATP synthesis as protons flow back through ATP synthase.
ATP synthase (Complex V) is one of the most elegant molecular machines in biology. It is a literal rotary motor — the F0 subunit embedded in the inner membrane forms a turbine, spun by the proton flow, that mechanically rotates the F1 catalytic head. Each 360-degree rotation of the rotor produces three ATP molecules through coordinated conformational changes at three catalytic sites. Each ATP requires the import of one molecule of inorganic phosphate from the cytoplasm and one molecule of ADP from cytoplasmic exchange.
Two critical transporters keep the ATP synthase fed:
- Adenine nucleotide translocase (ANT) — an antiporter in the inner mitochondrial membrane that exchanges newly synthesized ATP for cytoplasmic ADP, ensuring the matrix has the ADP it needs and the cytoplasm gets the ATP it needs.
- Phosphate carrier (PiC) — a symporter that imports inorganic phosphate together with a proton (the proton import dissipating a small fraction of the gradient, the cost of feeding ATP synthase its second substrate).
The system is exquisitely sensitive to the phosphate supply. When cytoplasmic phosphate concentration drops — whether from severe dietary deficiency, the rapid intracellular shift of refeeding syndrome, or the use of phosphate-binding antacids — the phosphate carrier delivers less phosphate to the matrix, ATP synthase operates below capacity, and oxidative phosphorylation rates fall. Tissues with the highest baseline energy demand (cardiac muscle, brain, skeletal muscle) feel this first.
Uncoupling is the deliberate or pathological dissipation of the proton gradient as heat rather than ATP synthesis. The brown-adipose-tissue protein UCP1 is the physiological example — it allows protons to leak back across the membrane through UCP1, dissipating the gradient as heat and generating non-shivering thermogenesis in newborns and cold-acclimated adults. The 1930s diet drug 2,4-dinitrophenol (DNP) is the toxicological example — it dramatically uncouples mitochondria, raising metabolic rate and core temperature, sometimes lethally. Both demonstrate that the tight coupling between phosphate utilization and energy capture is not automatic; it depends on the integrity of the inner mitochondrial membrane.
Phosphorus in Coenzymes and Metabolic Pathways
Beyond ATP itself, phosphorus is incorporated into nearly every coenzyme that drives intermediary metabolism. Without phosphate, the entire B-vitamin coenzyme machinery shuts down.
- NAD+ / NADH and NADP+ / NADPH — the universal redox cofactors. NAD+ accepts electrons in catabolic pathways (glycolysis, the citric acid cycle, fatty acid oxidation), and NADH delivers them to the electron transport chain. NADP+ supports anabolic reactions (fatty acid synthesis, cholesterol synthesis, nucleotide reduction) and antioxidant defense (regenerating reduced glutathione). Both molecules contain two phosphate groups in their dinucleotide backbone; NADP+ has an additional phosphate at the 2' position that distinguishes it.
- FAD / FADH2 and FMN / FMNH2 — the flavin coenzymes, both phosphorylated forms of riboflavin (vitamin B2). FAD is the prosthetic group of succinate dehydrogenase (Complex II of the electron transport chain) and of acyl-CoA dehydrogenases in fatty acid beta-oxidation.
- Coenzyme A (CoA) — built around a phosphopantetheine group with three phosphate atoms in its structure. CoA is the universal carrier of acyl groups in fatty acid oxidation, the citric acid cycle (acetyl-CoA, succinyl-CoA), and biosynthetic acetylation reactions (histone acetylation, acetylcholine synthesis).
- Cyclic AMP (cAMP) and cyclic GMP (cGMP) — second messengers built from a single phosphate cyclized between the 3' and 5' positions of the ribose sugar. cAMP mediates the intracellular effects of epinephrine, glucagon, ACTH, and many other hormones, linking phosphorus metabolism directly to hormonal regulation of energy balance.
- Pyridoxal-5'-phosphate (PLP) — the active coenzyme form of vitamin B6, essential for over 140 enzymatic reactions including amino acid transamination, decarboxylation, and neurotransmitter synthesis. See our Vitamin B6 page for more.
- Thiamine pyrophosphate (TPP) — the active form of vitamin B1, essential cofactor for pyruvate dehydrogenase, alpha-ketoglutarate dehydrogenase, and transketolase. Thiamine deficiency presents as beriberi and Wernicke encephalopathy precisely because the carbohydrate-metabolism pathways that require TPP cannot run.
The dependency chain is total: phosphate is required to synthesize ATP, ATP is required to phosphorylate B vitamins into their coenzyme forms, and those coenzymes are required to keep the central energy-producing pathways running. Lose phosphate, and the entire cascade unravels within hours.
Hypophosphatemia — When ATP Synthesis Fails
Hypophosphatemia is the clinical syndrome that results when serum inorganic phosphate falls below the normal range (typically 2.5 to 4.5 mg/dL in adults). The condition is graded by severity:
- Mild hypophosphatemia (2.0–2.5 mg/dL) — often asymptomatic; common in critical care, malnutrition, and post-major-surgery patients
- Moderate hypophosphatemia (1.0–2.0 mg/dL) — muscle weakness, fatigue, paresthesias; appears in chronic alcoholism, malabsorption, and the late stages of diabetic ketoacidosis treatment
- Severe hypophosphatemia (<1.0 mg/dL) — medical emergency. Manifestations include acute rhabdomyolysis, respiratory muscle failure requiring ventilatory support, cardiac dysfunction with depressed ejection fraction, hemolytic anemia from ATP depletion in erythrocytes (which lack mitochondria and depend entirely on glycolytic ATP), seizures, and coma.
The mechanistic chain is straightforward. Phosphate is needed for the GAPDH step of glycolysis and for ATP synthase in oxidative phosphorylation. When extracellular and intracellular phosphate fall together, both pathways slow. Tissues with high baseline ATP turnover — cardiac muscle, diaphragm, brain, kidney — experience an ATP deficit. Cell membrane sodium-potassium-ATPase fails, intracellular ions go awry, and cell death follows.
Erythrocytes are particularly affected because they have no mitochondria at all and depend entirely on glycolytic ATP to maintain membrane integrity and ion balance, plus on 2,3-bisphosphoglycerate (2,3-BPG) for normal oxygen unloading. In severe hypophosphatemia, red cell ATP and 2,3-BPG both fall, the oxygen dissociation curve shifts left (impairing oxygen delivery to tissues that need it), and red cells become rigid and lyse — the hemolytic anemia of severe hypophosphatemia.
Common causes:
- Refeeding syndrome — the single most common cause in hospitalized patients; see next section
- Diabetic ketoacidosis treatment — insulin therapy drives phosphate into cells along with glucose and potassium
- Chronic alcoholism — combined poor intake, malabsorption, urinary phosphate wasting, and thiamine deficiency
- Severe burns — massive phosphate losses through wound exudate plus catabolic demand
- Sepsis and critical illness — multifactorial; intracellular shift, urinary loss, and reduced intake
- Phosphate-binding antacids (aluminum hydroxide, calcium carbonate, sevelamer) used chronically without monitoring
- Hyperparathyroidism — PTH-driven renal phosphate wasting
- Genetic phosphate-wasting disorders — X-linked hypophosphatemia (XLH), autosomal dominant hypophosphatemic rickets (ADHR), and tumor-induced osteomalacia, all of which are driven by excess FGF23 signaling
Refeeding Syndrome and the Phosphate Shift
Refeeding syndrome is the textbook example of how rapidly phosphorus deficiency can become life-threatening. The setup is consistent: a chronically malnourished patient (severe anorexia nervosa, prolonged starvation, alcoholic ketoacidosis, post-bariatric surgery dehiscence, prolonged ICU stay with inadequate nutrition) has depleted total-body phosphate stores but maintains a roughly normal serum phosphate concentration because cellular metabolism has been ratcheted down and there is little phosphate demand.
When carbohydrate-rich nutrition is suddenly resumed — whether oral, enteral, or parenteral — insulin secretion surges. Insulin drives glucose into cells, where hexokinase immediately phosphorylates it to glucose-6-phosphate, consuming intracellular phosphate. Insulin also stimulates intracellular shifts of potassium and magnesium. The intracellular phosphate pool is rapidly depleted, the cell pulls phosphate from the extracellular fluid to compensate, and serum phosphate concentration drops precipitously — sometimes from a normal baseline to severe (<1.0 mg/dL) hypophosphatemia within 24 to 72 hours.
The consequences are catastrophic if not anticipated:
- Cardiac arrhythmias and sudden death — the most feared outcome; primarily mediated by hypophosphatemia plus hypokalemia plus hypomagnesemia all hitting cardiac conduction simultaneously
- Acute respiratory failure — diaphragm ATP depletion in patients who may already have marginal pulmonary reserve
- Rhabdomyolysis with secondary acute kidney injury
- Hemolytic anemia — the same erythrocyte ATP depletion mechanism described above
- Seizures, confusion, weakness — central and peripheral nervous system manifestations
- Wernicke encephalopathy — if thiamine is not co-supplemented; thiamine is a cofactor for several glycolytic enzymes and its deficiency is unmasked by the metabolic ramp-up
Modern protocols (the NICE guideline in the UK is the most widely cited) recommend identifying high-risk patients before refeeding, starting nutrition at no more than 5 to 10 kcal/kg/day for the first day or two, supplementing phosphate, potassium, magnesium, and thiamine prophylactically, and monitoring electrolytes every 12 hours during the first three to seven days of repletion. With this protocol, refeeding-syndrome mortality has dropped dramatically.
Clinical Applications and Repletion Strategies
For most adult patients in the developed world, dietary phosphorus intake exceeds requirements. The RDA for adults is 700 mg per day, and a typical Western diet delivers 1,000 to 1,800 mg, primarily from dairy, meat, fish, eggs, legumes, and whole grains, plus a significant additional contribution from inorganic phosphate additives in processed foods and soft drinks. The relevant clinical contexts where phosphorus repletion or restriction matters are concentrated:
- Refeeding-syndrome prophylaxis — oral or IV sodium or potassium phosphate at the time of nutritional repletion in any malnourished patient; protocol depends on severity but typically 0.3–0.6 mmol/kg/day for the first several days
- Critical care — routine measurement and repletion of serum phosphate in ICU patients; severe hypophosphatemia (<1.0 mg/dL) treated with IV sodium phosphate 15 mmol over 2–6 hours, repeated as needed
- Diabetic ketoacidosis — routine phosphate repletion is no longer recommended in mild-to-moderate DKA, but is indicated if serum phosphate falls below 1.0 mg/dL or if there is respiratory or cardiac dysfunction
- Chronic kidney disease — the opposite problem; restricted phosphate intake plus oral phosphate binders (calcium acetate, sevelamer, lanthanum) to control hyperphosphatemia, which drives secondary hyperparathyroidism and vascular calcification
- X-linked hypophosphatemia (XLH) — oral phosphate plus active vitamin D historically; burosumab (a monoclonal antibody against FGF23) is now the preferred first-line therapy
- Athletic performance — some evidence that creatine monohydrate supplementation (3–5 g/day) expands the muscle phosphocreatine pool by 10–20 percent in responders, modestly improving repeated-sprint and resistance-training performance; see our Creatine page for the supplementation evidence in detail
The asymmetry is striking: too little phosphate kills, but so does too much when the kidneys cannot clear it. The healthy kidney is the master regulator of phosphate balance through tightly tuned PTH and FGF23 signaling; when that regulation fails (CKD), phosphate accumulates and drives vascular calcification, secondary hyperparathyroidism, and the bone disease of renal osteodystrophy. The same nutrient that powers every cell becomes a cardiovascular toxin.
Key Research Papers
- Boyer PD (1997). The ATP synthase — a splendid molecular machine. Annual Review of Biochemistry. — PubMed
- Walker JE (2013). The ATP synthase: the understood, the uncertain and the unknown. Biochemical Society Transactions. — PubMed
- Mitchell P (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. — PubMed
- Wallimann T et al. (1992). Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the 'phosphocreatine circuit'. Biochemical Journal. — PubMed
- Hue L, Rider MH (1987). Role of fructose 2,6-bisphosphate in the control of glycolysis in mammalian tissues. Biochemical Journal. — PubMed
- Subramanian R, Khardori R (2000). Severe hypophosphatemia: pathophysiologic implications, clinical presentations, and treatment. Medicine. — PubMed
- Mehanna HM et al. (2008). Refeeding syndrome: what it is, and how to prevent and treat it. BMJ. — PubMed
- Friedli N et al. (2018). Management and prevention of refeeding syndrome in medical inpatients: An evidence-based and consensus-supported algorithm. Nutrition. — PubMed
- Brosnan ME, Brosnan JT (2016). The role of dietary creatine. Amino Acids. — PubMed
- Hultman E et al. (1996). Muscle creatine loading in men. Journal of Applied Physiology. — PubMed
- Knochel JP (1977). The pathophysiology and clinical characteristics of severe hypophosphatemia. Archives of Internal Medicine. — PubMed
- Hardie DG (2014). AMPK — sensing energy while talking to other signaling pathways. Cell Metabolism. — PubMed
PubMed Topic Searches
- PubMed: ATP synthase structure and function
- PubMed: Phosphocreatine and energy metabolism
- PubMed: Hypophosphatemia and refeeding
- PubMed: Oxidative phosphorylation
- PubMed: Glycolysis and ATP production
Connections
- Phosphorus Overview
- Phosphorus Benefits Hub
- Phosphorus for Bone Mineralization
- Phosphorus for Cell Membranes
- Phosphorus for Acid-Base Balance
- Creatine
- Magnesium (ATP Cofactor)
- Calcium
- Potassium
- Vitamin B6 (PLP Coenzyme)
- Vitamin B1 (Thiamine / TPP)
- Vitamin D3
- Kidney Disease
- Anemia
- Osteoporosis
- eGFR
- Eggs
- All Minerals