Phosphorus for Acid-Base Balance
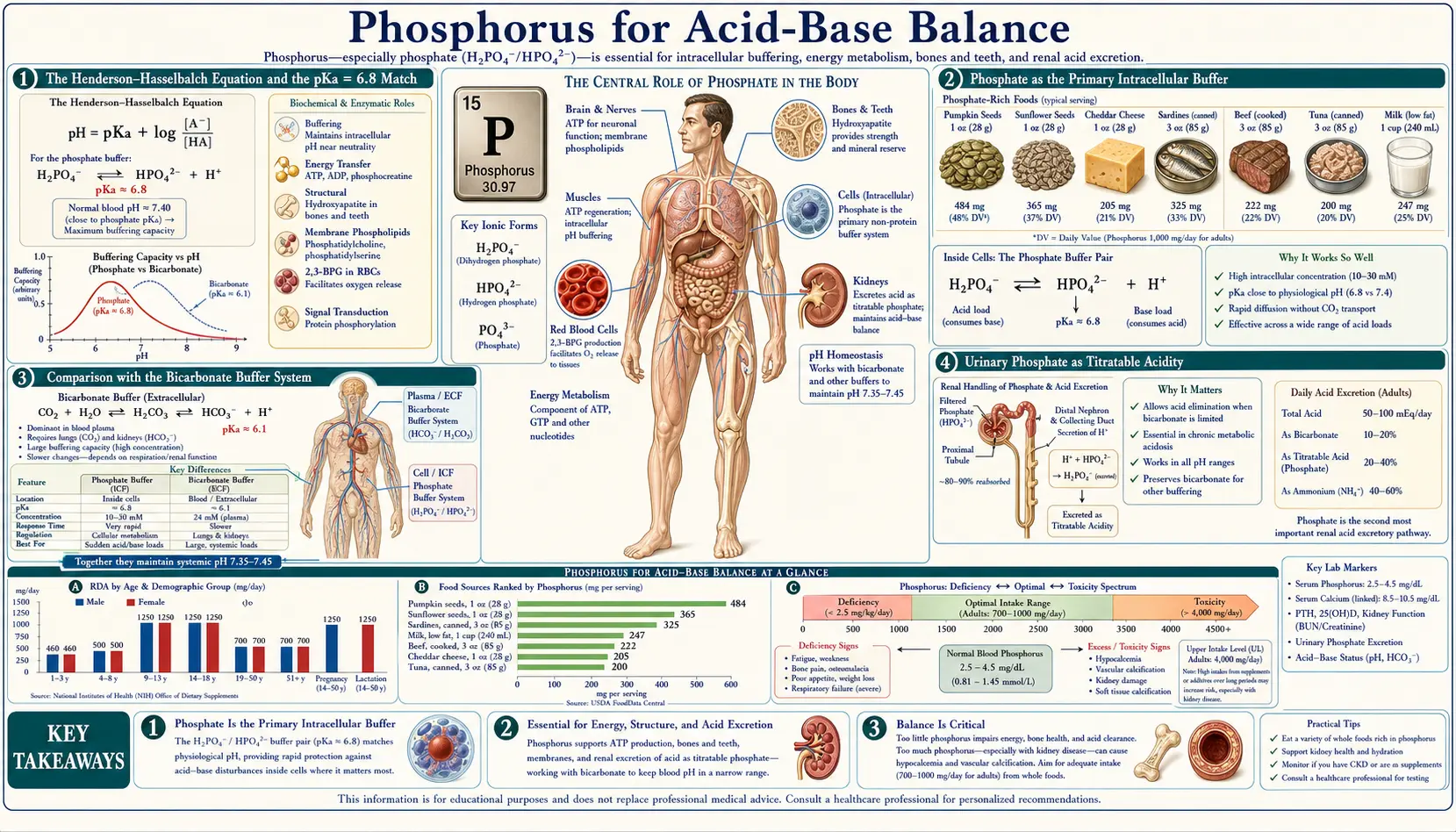
Phosphate is the primary intracellular buffer in human biology. Its dihydrogen-hydrogen pair, H2PO4− ↔ HPO42−, has a pKa of 6.8 — remarkably close to the intracellular pH of 7.0 to 7.2, which makes it the most thermodynamically effective buffer in the cytoplasm and the lysosome. In the urine, the same phosphate equilibrium accounts for the "titratable acidity" that drives daily renal excretion of fixed dietary acid. In diabetic ketoacidosis, the depletion of intracellular phosphate during prolonged ketosis becomes a major contributor to the rebound hypophosphatemia of insulin therapy. And in renal tubular acidosis, the phosphate buffer system is one of the kidney's key levers for restoring extracellular bicarbonate. This deep-dive walks through the biochemistry of the phosphate buffer, the kidney's use of urinary phosphate to excrete metabolic acid, the special case of DKA, and the dangerous misuses of phosphate-based enemas in vulnerable populations.
Table of Contents
- The Henderson-Hasselbalch Equation and the pKa = 6.8 Match
- Phosphate as the Primary Intracellular Buffer
- Comparison with the Bicarbonate Buffer System
- Urinary Phosphate as Titratable Acidity
- Daily Renal Acid Excretion — the Numbers
- Phosphate in Diabetic Ketoacidosis
- Metabolic Acidosis and Compensation
- Renal Tubular Acidosis and the Phosphate Buffer
- Phosphate Enemas and Acid-Base Disturbance
- Clinical Applications and Lab Interpretation
- Key Research Papers
- Connections
- Featured Videos
The Henderson-Hasselbalch Equation and the pKa = 6.8 Match
The Henderson-Hasselbalch equation describes the pH of any buffered solution in terms of the pKa of the buffer and the ratio of the conjugate base to the conjugate acid:
pH = pKa + log10([base] / [acid])
A buffer is most effective at resisting pH changes when the pH is within one unit of the pKa — that is, when the ratio of base to acid is between 1:10 and 10:1. The maximum buffering capacity is at pH = pKa, where the ratio is exactly 1:1 and the buffer is half-protonated. The further the pH drifts from the pKa, the worse the buffering performance becomes.
For the phosphate equilibrium H2PO4− ↔ H+ + HPO42−, the pKa is 6.8. Intracellular pH is 7.0 to 7.2. The ratio [HPO42−] / [H2PO4−] is therefore approximately 1.6 to 2.5 — well within the optimal buffering range. Compare this with bicarbonate, where the pKa for the carbonic-acid / bicarbonate equilibrium (after accounting for CO2 hydration) is approximately 6.1, well below intracellular pH; the bicarbonate base-to-acid ratio is approximately 10:1, at the edge of effective buffering. Phosphate is intrinsically better matched to intracellular pH than any other physiologically abundant buffer.
The same principle applies inside lysosomes. Lysosomal pH is approximately 4.5 to 5.0 — far from the phosphate pKa — so phosphate is a poor buffer there, and the lysosome relies instead on the acidic side chains of luminal proteins for buffering capacity. Phosphate dominates wherever pH is near 6.8, and other buffers dominate elsewhere.
Phosphate as the Primary Intracellular Buffer
The intracellular fluid contains roughly 100 mEq/L of phosphate (across all its forms — inorganic phosphate plus the phosphate in ATP, organic phosphate compounds, and phospholipids). Compare this with bicarbonate at approximately 10 to 15 mEq/L in the cytoplasm (much lower than the 24 mEq/L of extracellular bicarbonate, because the cytoplasm is at lower pH and shifts toward CO2). The combination of high intracellular concentration plus the favorable pKa makes phosphate the dominant intracellular buffer by a substantial margin.
The intracellular phosphate buffer is loaded with hydrogen ions from several continuous metabolic sources:
- ATP hydrolysis — releases a small net hydrogen ion at physiological pH (the products ADP and Pi are slightly more acidic than ATP)
- Glycolysis — produces lactate plus a proton in oxygen-limited conditions; even with mitochondrial pyruvate oxidation, the net glycolytic flux generates protons
- Beta-oxidation of fatty acids — produces acetyl-CoA, which under normal conditions enters the citric acid cycle but under conditions of glucose excess or insulin resistance can divert toward ketone body formation
- Ammonium production — from amino acid catabolism, particularly glutamine breakdown in renal tubular cells
- Carbonic acid — CO2 from oxidative metabolism combines with water to form H2CO3, which dissociates to bicarbonate and a proton
The phosphate buffer absorbs these protons as they appear, converting HPO42− to H2PO4− and keeping intracellular pH within narrow bounds. The cell's membrane sodium-hydrogen exchanger (NHE1) and other ion transporters then export the accumulated protons across the plasma membrane to the extracellular fluid, where the bicarbonate buffer absorbs them and the lung disposes of the resulting CO2. The kidney handles the small daily net excess of fixed acid (see the titratable acidity section below).
Lose intracellular phosphate — through chronic hypophosphatemia, refeeding-syndrome phosphate shifts, severe hyperventilation that depletes intracellular phosphate as cellular phosphorylation runs forward — and intracellular pH becomes more vulnerable to swing. The clinical correlate is that severe hypophosphatemia patients are at increased risk for both lactic acidosis and ketoacidosis because the buffering reserve is depleted.
Comparison with the Bicarbonate Buffer System
The body uses several buffer systems in parallel; each has a different domain of effectiveness:
- Bicarbonate / carbonic acid (HCO3− / H2CO3) — the dominant extracellular buffer. Effective pKa of approximately 6.1, but the system is open (the lung can blow off CO2, increasing the apparent buffering capacity dramatically). Accounts for approximately 50 percent of the body's total buffering capacity.
- Phosphate (H2PO4− / HPO42−) — the dominant intracellular buffer. pKa 6.8, optimally matched to intracellular pH. Accounts for approximately 5 percent of overall body buffering and a much larger fraction of intracellular buffering specifically.
- Hemoglobin and plasma proteins — the imidazole side chains of histidine residues have pKa values near 6.5 to 7.0, making proteins (especially the abundant hemoglobin in red cells) effective intracellular and intravascular buffers. Accounts for approximately 35 percent of overall body buffering.
- Bone hydroxyapatite — releases calcium and phosphate during chronic acidosis, effectively buffering at the cost of bone mineral. Slow but important in chronic metabolic acidosis (renal failure, chronic vegetarianism with high acid load, type IV renal tubular acidosis).
The phosphate buffer is unique among these in being intrinsically suited to intracellular pH. The bicarbonate system is far more powerful in the extracellular space (because the lung can rapidly dispose of CO2, effectively removing the conjugate acid), but in the cytoplasm where there is no rapid CO2 sink, phosphate dominates. The two systems work in series — phosphate handles the moment-to-moment intracellular swings, then the cell exports protons to the extracellular fluid where the bicarbonate buffer takes over, and the kidney handles the slow net daily acid load.
Urinary Phosphate as Titratable Acidity
The kidney must dispose of approximately 50 to 100 mEq of fixed acid per day in a typical Western diet (the "dietary acid load" from sulfur-containing amino acids in animal protein, plus other organic acids). This acid cannot be excreted as bicarbonate (the kidney has just absorbed all of the filtered bicarbonate to maintain extracellular pH); it must be excreted as hydrogen ion bound to some urinary buffer that can carry the proton out without dropping urinary pH below the minimum the renal tubule can tolerate (approximately pH 4.5).
Two urinary buffers do the work:
- Titratable acidity (phosphate) — filtered phosphate in the renal tubule accepts secreted hydrogen ions, converting HPO42− to H2PO4−. This accounts for approximately one-third of daily acid excretion in a typical diet, equivalent to roughly 20 to 40 mEq/day of acid load.
- Ammonium excretion — renal tubular cells synthesize ammonia from glutamine, which traps a hydrogen ion in the tubular lumen as ammonium (NH4+). This accounts for the remaining two-thirds of daily acid excretion and can be massively upregulated during chronic metabolic acidosis — ammonium excretion can rise from a baseline of approximately 40 mEq/day to over 250 mEq/day during sustained acidosis.
The phosphate contribution is capped by the filtered phosphate load (typically 1,000 to 1,500 mg/day, equivalent to roughly 33 to 50 mmol). Each phosphate molecule can accept one proton at urinary pH 4.5 to 5.0 (where the H2PO4− form dominates). This sets the ceiling on titratable acidity at roughly 50 mEq/day. The ammonium system can be expanded; the phosphate system cannot. For sustained metabolic acidosis (chronic kidney disease, ketoacidosis), ammonium excretion is the dominant adaptive response and titratable acidity remains relatively constant.
Daily Renal Acid Excretion — the Numbers
For a typical adult eating a standard Western mixed-protein diet, the daily net acid production is approximately 50 to 70 mEq. This comes from:
- Sulfur-containing amino acids (methionine, cysteine) metabolized to sulfuric acid — the largest single source, approximately 30 to 50 mEq/day in a high-animal-protein diet
- Phosphate ester hydrolysis from dietary phosphoproteins and phospholipids — approximately 10 to 20 mEq/day
- Organic acids (lactate, citrate, other intermediates) that are normally fully metabolized to CO2 and water but a small net amount escapes as fixed acid
- Incomplete oxidation of glucose under conditions of brief tissue hypoxia or vigorous exercise — small but persistent contribution
This net acid is buffered in the moment by the extracellular bicarbonate, then handled by the kidney over the following hours. The kidney's response:
- Reabsorbs 100 percent of filtered bicarbonate — approximately 4,400 mEq/day (assuming GFR 180 L/day × 24 mEq/L). The proximal tubule does ~80 percent of this work, the thick ascending limb of Henle does most of the rest. None of this counts as net acid excretion; it is bicarbonate conservation.
- Excretes titratable acidity as urinary H2PO4− — approximately 20 to 40 mEq/day
- Excretes ammonium as urinary NH4+ — approximately 30 to 50 mEq/day
- Generates new bicarbonate — one new bicarbonate is generated and added to the venous blood for every proton excreted in the urine (whether bound to phosphate or to ammonium), restoring the extracellular bicarbonate pool consumed by the daily acid load
The arithmetic works out: net acid production approximately equals titratable acidity plus ammonium excretion minus any bicarbonaturia. In chronic kidney disease, as renal mass falls, the ability to excrete ammonium falls (the proximal-tubule glutaminase machinery is concentrated in nephrons), and the chronic metabolic acidosis of CKD develops. The phosphate-bound titratable acidity component holds up relatively better than the ammonium component because it is a passive process driven by filtered load.
Phosphate in Diabetic Ketoacidosis
Diabetic ketoacidosis (DKA) is a striking example of the interconnection between phosphate, intracellular pH, and overall acid-base balance. The pathophysiology starts with insulin deficiency (whether absolute in type 1 diabetes or relative in type 2 under severe stress): unrestrained lipolysis releases free fatty acids, the liver oxidizes them to acetyl-CoA, and the acetyl-CoA pours into ketone body production (acetoacetate, beta-hydroxybutyrate, acetone) faster than peripheral tissues can use it.
The result is a severe anion-gap metabolic acidosis — blood pH often 7.0 to 7.2, bicarbonate often less than 10 mEq/L, anion gap often greater than 20 mEq/L. Simultaneously, profound osmotic diuresis from hyperglycemia depletes total-body water, sodium, potassium, magnesium, and phosphate — even though serum potassium and serum phosphate may appear normal or even elevated initially because of the intracellular-to-extracellular shift of these ions under acidotic conditions and insulin lack.
Insulin therapy reverses the picture rapidly. Glucose enters cells, is phosphorylated, and the intracellular machinery ramps up its phosphate consumption. Potassium and phosphate both shift back into cells with the insulin and glucose. Serum potassium and serum phosphate both drop, often precipitously. This is where the rebound hypophosphatemia of DKA treatment becomes clinically important:
- Serum phosphate at presentation is often normal or elevated
- Within 4 to 12 hours of insulin therapy, serum phosphate can fall to severe levels (<1.0 mg/dL)
- Severe hypophosphatemia in this setting can cause respiratory muscle weakness (impairing ventilation in a patient who needs to blow off the CO2 generated by bicarbonate buffering), hemolysis, rhabdomyolysis, and cardiac dysfunction
Modern DKA management guidelines (ADA, NICE) recommend monitoring serum phosphate every 2 to 4 hours during the first 24 hours of insulin therapy and supplementing phosphate (as potassium phosphate, taking advantage of the simultaneous potassium need) if serum phosphate falls below approximately 1.0 mg/dL or if there is respiratory or cardiac compromise. Routine empirical phosphate replacement in mild-to-moderate DKA has not been shown to improve outcomes and adds complexity, but targeted replacement in severe cases is standard of care.
Metabolic Acidosis and Compensation
Metabolic acidosis is the clinical syndrome of decreased extracellular pH driven by either accumulation of fixed acid (lactic acidosis, ketoacidosis, uremic acidosis) or loss of bicarbonate (diarrhea, type 2 renal tubular acidosis). The compensatory responses involve all the buffer systems described above, plus respiratory and renal adjustments.
The compensation cascade:
- Immediate (seconds to minutes) — extracellular bicarbonate buffers the new acid load, decreasing serum bicarbonate. Intracellular phosphate and protein buffers absorb a smaller fraction of the proton load.
- Respiratory (minutes to hours) — the carotid and aortic chemoreceptors detect the falling pH and increase ventilation, lowering arterial CO2 and pulling the bicarbonate equilibrium back toward bicarbonate. Each 1 mEq/L drop in serum bicarbonate is approximately compensated by a 1.2 mmHg drop in arterial PCO2 (Winters formula). Kussmaul breathing in DKA is the visible expression of this response.
- Renal (hours to days) — the kidney upregulates ammoniagenesis (raising urinary NH4+ excretion from baseline 30–50 mEq/day toward 200+ mEq/day) and accelerates net new bicarbonate generation. Urinary phosphate-bound titratable acidity stays relatively constant unless dietary phosphate intake changes; the kidney does not have a major mechanism for upregulating phosphate-bound acid excretion.
- Skeletal (days to months in chronic acidosis) — bone hydroxyapatite slowly dissolves, releasing calcium and phosphate. The calcium consumes the excess proton load and the phosphate then contributes additional urinary titratable acidity. Over years, this is one of the mechanisms behind the bone disease of chronic kidney disease and chronic dietary acid excess.
The interplay matters clinically. Patients with chronic metabolic acidosis from CKD develop progressive bone disease partly because of this slow-burning bone-dissolution buffering. Modern CKD guidelines therefore recommend oral sodium bicarbonate supplementation to maintain serum bicarbonate above 22 mEq/L, both for symptom control and for skeletal preservation.
Renal Tubular Acidosis and the Phosphate Buffer
Renal tubular acidosis (RTA) is a family of disorders in which the kidney fails to maintain normal acid-base balance despite a normal glomerular filtration rate. The kidney's ability to use phosphate as a urinary buffer is the lever distinguishing some forms from others:
- Type 1 (distal) RTA — the alpha-intercalated cells of the cortical collecting duct cannot secrete protons effectively. Urinary pH is inappropriately alkaline (>5.5) despite systemic acidosis. Titratable acidity is low because the phosphate that reaches the collecting duct cannot be protonated to H2PO4− if the tubule cannot secrete the proton. Ammonium excretion is also impaired. Causes include genetic defects in the V-type proton ATPase, autoimmune disease (Sjögren syndrome is classic), and several medications (amphotericin B, lithium).
- Type 2 (proximal) RTA — the proximal tubule fails to reabsorb the filtered bicarbonate, dumping it into the urine. Serum bicarbonate falls to a new steady-state level (typically 12 to 18 mEq/L), at which the reduced filtered load equals the impaired reabsorptive capacity, and below that level the distal tubule recovers normal acidification ability. Urinary pH is variable. Causes include Fanconi syndrome (often with associated phosphaturia), heavy metals, and several inherited disorders.
- Type 4 (hyperkalemic) RTA — aldosterone deficiency or resistance, with impaired ammoniagenesis. This is the most common form encountered in adult clinical practice, especially in diabetic nephropathy with hyporeninemic hypoaldosteronism. The phosphate-bound titratable acidity component is relatively preserved; the ammonium component fails.
The diagnostic value of the urinary anion gap (Na+ + K+ − Cl−, a surrogate for urinary ammonium when the gap is negative) and the urinary osmolar gap (a more direct surrogate) hinges on the principle that the phosphate-bound titratable acidity component is relatively constant across individuals (because it's set by dietary phosphate intake) while the ammonium component is what varies. In acidosis with intact ammoniagenesis, urinary ammonium is high and the urinary anion gap is markedly negative (less than −40). In acidosis with impaired ammoniagenesis (type 1 RTA, type 4 RTA), urinary ammonium is low and the urinary anion gap is positive or only mildly negative.
Phosphate Enemas and Acid-Base Disturbance
Warning — sodium phosphate enemas in vulnerable populations. Phosphate-based enemas (Fleet enemas, sodium phosphate enemas) are a common over-the-counter and prescription bowel preparation. In healthy adults with normal kidney function used as directed, they are generally safe. In several vulnerable populations — the elderly, patients with reduced kidney function, patients on ACE inhibitors or ARBs, patients with bowel obstruction or perforation, children — they can cause life-threatening hyperphosphatemia, hypocalcemia, hypernatremia, hypokalemia, and acute kidney injury. The FDA issued a safety alert in 2014 highlighting case reports of fatal outcomes when phosphate enemas were used in elderly patients with bowel obstruction or impaired kidney function.
The mechanism is straightforward. A typical sodium phosphate enema contains approximately 19 grams of monobasic and dibasic sodium phosphate, delivering roughly 5 grams of elemental phosphorus. If the bowel is obstructed and the phosphate solution cannot be eliminated, the phosphate is absorbed across the colonic mucosa at rates far exceeding the kidney's ability to clear it. Serum phosphate can rise to 20 mg/dL or higher (normal range 2.5–4.5). The phosphate then complexes with serum calcium (forming insoluble calcium phosphate precipitates), driving down ionized calcium to dangerous levels and depositing calcium phosphate in soft tissue including the renal tubules (acute phosphate nephropathy). Cardiac arrhythmias from hypocalcemia and hyperphosphatemia, plus acute kidney injury from precipitated calcium phosphate, account for the worst outcomes.
Safer alternatives for bowel preparation in vulnerable populations include polyethylene glycol (PEG) lavage, which is osmotically inert and not absorbed, and tap water enemas. The clinical lesson: phosphate has narrow safety margins outside the body's normal regulatory range, and the same molecule that powers every cell can become acutely toxic when delivered in pharmacologic excess to a patient who cannot clear it.
Clinical Applications and Lab Interpretation
- Suspected metabolic acidosis — arterial blood gas (pH, PCO2, calculated bicarbonate), serum chemistry panel (sodium, potassium, chloride, bicarbonate, BUN, creatinine, glucose), serum lactate, ketones. Calculate the anion gap (Na+ − Cl− − HCO3−; normal 8–12); a wide gap points to organic acid accumulation (lactate, ketones, toxic alcohols, uremia).
- Suspected RTA — first confirm metabolic acidosis with a normal anion gap (hyperchloremic). Then check urine pH, urinary anion gap, serum potassium. Type 1 RTA shows urine pH >5.5 despite systemic acidosis; type 2 RTA shows variable urine pH with proximal-tubule features (glucosuria, aminoaciduria, phosphaturia); type 4 RTA shows hyperkalemia plus normal urine pH.
- DKA — standard treatment is IV fluids, insulin infusion, potassium replacement once urine output is established, and serial monitoring of serum potassium, phosphate, calcium, magnesium, glucose, and bicarbonate every 2 to 4 hours during the first 24 hours.
- Chronic metabolic acidosis of CKD — aim for serum bicarbonate above 22 mEq/L; oral sodium bicarbonate 600 to 1,300 mg three times daily is typical; correction has been shown in trials to slow CKD progression and preserve muscle mass.
- Phosphate-based bowel prep — review contraindications carefully (age >55, CKD, ACE inhibitor / ARB / NSAID use, dehydration, electrolyte imbalance, congestive heart failure, bowel obstruction). Substitute PEG lavage in any patient with even relative contraindications. Never use in suspected bowel obstruction.
- 24-hour urine workup — for recurrent calcium kidney stones, evaluation of hypercalciuria, or assessment of acid-base status. Includes urinary phosphate, urinary calcium, citrate, oxalate, sodium, potassium, ammonium, sulfate, and creatinine.
For interpretation of related labs, see our eGFR page and the broader Kidney Function Tests page.
Key Research Papers
- Hamm LL et al. (2015). Acid-base homeostasis. Clinical Journal of the American Society of Nephrology. — PubMed
- Halperin ML, Goldstein MB (2010). Fluid, Electrolyte, and Acid-Base Physiology: A Problem-Based Approach. Saunders. — PubMed
- Adrogué HJ, Madias NE (1998). Management of life-threatening acid-base disorders. NEJM. — PubMed
- Kitabchi AE et al. (2009). Hyperglycemic crises in adult patients with diabetes. Diabetes Care. — PubMed
- Fisher JN, Kitabchi AE (1983). A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. JCEM. — PubMed
- de Brito-Ashurst I et al. (2009). Bicarbonate supplementation slows progression of CKD and improves nutritional status. JASN. — PubMed
- Markowitz GS, Perazella MA (2009). Acute phosphate nephropathy. Kidney International. — PubMed
- Mehta RL et al. (2014). Phosphate management in chronic kidney disease. KDIGO Controversies Conference report. — PubMed
- DuBose TD Jr (1997). Hyperkalemic hyperchloremic metabolic acidosis: pathophysiologic insights. Kidney International. — PubMed
- Rodriguez Soriano J (2002). Renal tubular acidosis: the clinical entity. JASN. — PubMed
- Lemann J Jr et al. (2003). Bone buffering of acid and base in humans. American Journal of Physiology — Renal Physiology. — PubMed
- Ori Y et al. (2012). Fatalities and severe metabolic disorders associated with the use of sodium phosphate enemas: a single center's experience. Archives of Internal Medicine. — PubMed
PubMed Topic Searches
- PubMed: Phosphate buffer and intracellular pH
- PubMed: Titratable acidity and urinary phosphate
- PubMed: DKA and phosphate
- PubMed: RTA diagnosis
- PubMed: Phosphate enema nephropathy
Connections
- Phosphorus Overview
- Phosphorus Benefits Hub
- Phosphorus for Energy Production
- Phosphorus for Bone Mineralization
- Phosphorus for Cell Membranes
- Potassium
- Magnesium
- Calcium
- Type 1 Diabetes
- Type 2 Diabetes
- Kidney Disease
- Pulmonology (Respiratory Compensation)
- eGFR
- Kidney Function Tests
- Vitamin D3
- Hyperparathyroidism
- All Minerals