Hypermobile EDS and the 2017 Diagnostic Criteria
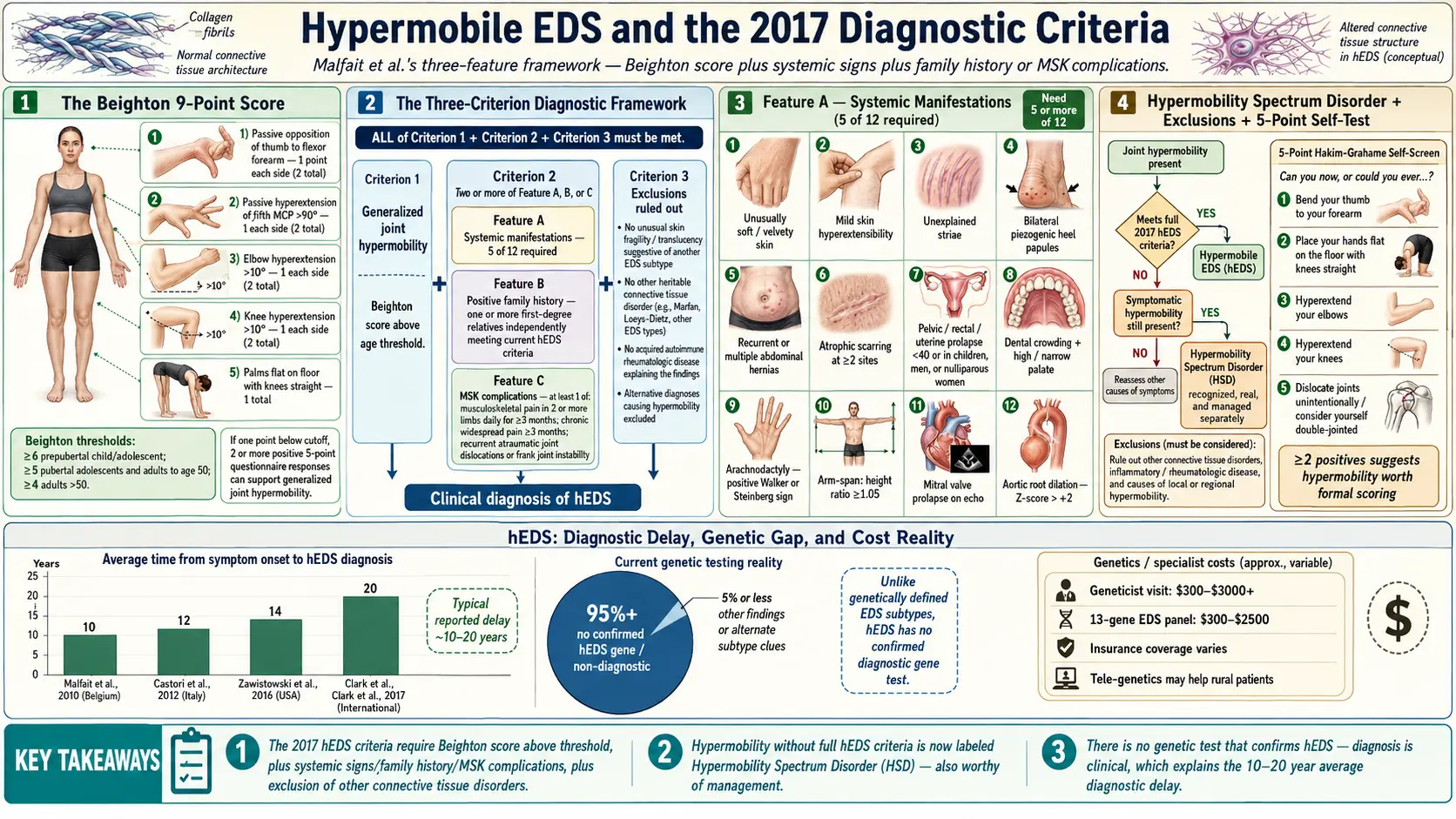
Table of Contents
- What Hypermobile EDS Is — and Why It Is Different
- The Beighton 9-Point Score
- Age- and Sex-Adjusted Beighton Thresholds
- Historical GJH vs. hEDS — The Shift in Terminology
- The 2017 hEDS Diagnostic Criteria (Malfait et al.)
- Feature A — Systemic Manifestations
- Feature B (Family History) and Feature C (MSK Complications)
- The Exclusion Criterion — Ruling Out Other Disorders
- Hypermobility Spectrum Disorder (HSD)
- The Criteria Controversy
- Self-Assessment at Home — The 5-Point Questionnaire
- Finding a Knowledgeable Provider
- What the Genetic Test Gap Means
- Classical EDS (cEDS) as a Contrast
- Cost Reality — Geneticist Visits and Panels
- Key Research Papers
- Connections
- Featured Videos
What Hypermobile EDS Is — and Why It Is Different
Hypermobile Ehlers-Danlos Syndrome (hEDS) is the most common subtype of EDS, accounting for an estimated 80–90% of all EDS cases. It is also the only one of the thirteen currently recognized EDS subtypes without an identified gene. Classical EDS has pathogenic variants in COL5A1 and COL5A2. Vascular EDS is tied to COL3A1. Kyphoscoliotic, arthrochalasia, dermatosparaxis, and the other rarer subtypes each have molecular signatures that a lab can confirm from a blood or saliva sample. hEDS does not — not yet.
That single fact shapes everything about how hEDS is diagnosed, doubted, studied, and funded. Without a gene, there is no confirmatory test. The diagnosis is made from the patient's body and history against a published clinical checklist. It is a diagnosis of pattern-recognition and exclusion, which puts enormous weight on the clinician seeing it and the criteria being used.
The current rulebook is the 2017 International Classification of the Ehlers-Danlos Syndromes, published by Malfait and colleagues, with a companion paper by Castori and colleagues explaining the framework. These two papers replaced the older 1998 Villefranche criteria and, deliberately, made hEDS harder to diagnose than it had been. The rationale was scientific — a tighter clinical definition would produce cleaner research cohorts, which would speed up the hunt for the gene — but the human cost has been substantial. Many patients with obviously loose joints, chronic pain, frequent subluxations, and a textbook family tree now fall outside the strict boundary.
The Beighton 9-Point Score
The Beighton score is a quick bedside test of generalized joint hypermobility (GJH). It scores nine maneuvers, one point each, for a maximum of 9:
- Passive hyperextension of the 5th finger beyond 90 degrees — 1 point per hand, 2 total.
- Passive apposition of the thumb to the flexor side of the forearm — 1 point per hand, 2 total.
- Active hyperextension of the elbow beyond 10 degrees — 1 point per elbow, 2 total.
- Active hyperextension of the knee beyond 10 degrees — 1 point per knee, 2 total.
- Forward flexion of the trunk with knees fully extended so that palms rest flat on the floor — 1 point (not bilateral).
A clinician should measure the elbow and knee hyperextension with a goniometer rather than eyeballing it — the 10-degree cutoff matters, and it is easy to over- or under-call without one. Many patients score the points they do not remember scoring; hypermobile people tend to assume their range of motion is normal because it is the only range they have ever had.
Age- and Sex-Adjusted Beighton Thresholds
The 2017 criteria recognize that joint laxity decreases with age. A 45-year-old who can no longer put her palms on the floor may have scored 8 out of 9 as a teenager. The thresholds for meeting the GJH requirement of hEDS are:
- Prepubertal children (roughly under 12–13): ≥6 out of 9.
- Pubertal through age 50: ≥5 out of 9.
- Over 50: ≥4 out of 9.
- Adult men at any age: the threshold is often functionally applied as ≥4 given that men score lower than women on average at every age.
If a patient's current Beighton is below threshold but history, photographs, or witness accounts support a higher score in youth, the clinician can use the 5-point questionnaire (see self-assessment below) as a historical supplement. Two or more "yes" answers count as evidence of GJH even in a now-stiffer adult body.
Historical GJH vs. hEDS — The Shift in Terminology
Before 2017, the field used a simpler hierarchy: benign joint hypermobility syndrome for people with loose joints and pain but no other systemic findings, and hEDS (then called EDS-hypermobility type or EDS Type III) for people with more body-wide involvement. The line between them was blurry, and the same patient could carry either label depending on which clinic she walked into.
The 2017 rewrite drew a sharper line. Generalized joint hypermobility (GJH) became an objective, measurable finding — a Beighton score above the age-adjusted threshold. hEDS became a specific multi-system syndrome with a checklist. Patients who fall between the two — hypermobile with symptoms but not quite meeting the full hEDS checklist — moved into a new umbrella called Hypermobility Spectrum Disorder (HSD).
The 2017 hEDS Diagnostic Criteria (Malfait et al.)
To receive an hEDS diagnosis under the current rules, a patient must meet all three of the following criteria simultaneously:
- Criterion 1 — Generalized joint hypermobility (GJH). Beighton score at or above the age-adjusted threshold, or — if below threshold — a combination of a borderline Beighton plus two or more "yes" answers on the 5-point questionnaire to establish historical GJH.
- Criterion 2 — Two or more of Feature A, Feature B, or Feature C (described below).
- Criterion 3 — Exclusion of other heritable and acquired connective tissue disorders.
All three must be satisfied. Missing any one of the three means the patient does not meet the hEDS definition, regardless of how hypermobile or how symptomatic she is.
Feature A — Systemic Manifestations
Feature A is a list of twelve body-wide signs of abnormal connective tissue. The patient must have at least five of them to count Feature A as "present." The twelve signs are:
- Unusually soft or velvety skin.
- Mild skin hyperextensibility (the skin lifts away from the forearm more than usual, but springs back).
- Unexplained striae (stretch marks) in places not related to weight gain or pregnancy, such as the back, thighs, or breasts.
- Bilateral piezogenic papules of the heel — small fat herniations that bulge out when standing.
- Recurrent or multiple abdominal hernias.
- Atrophic scarring in at least two sites, without the wide, papery "cigarette-paper" scars that define classical EDS.
- Pelvic floor, rectal, or uterine prolapse in a patient without obvious risk factors (e.g., no multiple vaginal deliveries, no long-term heavy lifting).
- Dental crowding and a high or narrow palate.
- Arachnodactyly — defined by a positive wrist sign (Walker), a positive thumb sign (Steinberg), or both.
- Arm-span-to-height ratio ≥1.05.
- Mitral valve prolapse (mild or greater) on echocardiogram.
- Aortic root dilatation with a Z-score >+2.
A pectus excavatum or carinatum is also flagged in the 2017 criteria as a supporting skeletal finding. Five of twelve is a fairly demanding bar, and many hypermobile patients who look obviously "connective-tissue different" score three or four.
Feature B (Family History) and Feature C (MSK Complications)
Feature B is simple: a positive family history, meaning one or more first-degree relatives (parent, full sibling, or child) independently meets the hEDS criteria. "Family members who seem bendy" does not count — the relative has to meet the full 2017 checklist. This is one of the most restrictive parts of the criteria, because in many families the oldest known affected member was diagnosed before 2017 under older rules, or was never evaluated at all.
Feature C is the musculoskeletal burden of disease. It is met by any of the following:
- Musculoskeletal pain in two or more limbs, recurring daily, for at least three months.
- Chronic widespread pain for three or more months.
- Recurrent joint dislocations or frank joint instability in the absence of trauma, defined as either three or more atraumatic dislocations in the same joint, or two or more atraumatic dislocations in two different joints occurring at different times.
Feature C is often the easiest for symptomatic adults to meet and the hardest for children, whose pain may be dismissed as "growing pains" and whose subluxations may not yet have happened enough times to count.
The Exclusion Criterion — Ruling Out Other Disorders
Criterion 3 requires the clinician to actively exclude other conditions that can mimic hEDS. At minimum, this means ruling out:
- Other EDS subtypes — especially classical EDS (COL5A1, COL5A2) and vascular EDS (COL3A1), which have specific genetic tests. A multi-gene connective-tissue panel is the standard way to exclude them in one sweep.
- Marfan syndrome (FBN1 variants), which shares the tall, long-limbed, high-palate phenotype but is driven by fibrillin-1 defects and carries an aortic-dissection risk profile of its own.
- Loeys-Dietz syndrome (LDS) — caused by variants in the TGF-β signaling pathway genes (TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3) — which also presents with hypermobility, skin findings, and vascular risk.
- Osteogenesis imperfecta in its milder forms (COL1A1, COL1A2), which can overlap symptomatically.
- Rheumatoid arthritis and other inflammatory arthritides, which can produce joint pain, swelling, and instability from a different mechanism.
- Acquired connective tissue disease such as lupus or mixed connective tissue disease, which can loosen joints through chronic inflammation rather than heritable collagen defects.
In practice, exclusion is done with a combination of a genetic panel, an echocardiogram to screen for aortic root dilatation, basic labs (ANA, CBC, inflammatory markers), and a careful clinical exam.
Hypermobility Spectrum Disorder (HSD)
Hypermobility Spectrum Disorder (HSD) is the diagnostic umbrella for hypermobile patients with clinical problems who do not meet the full 2017 hEDS checklist. It was introduced in 2017 specifically to catch this group. HSD has four recognized variants:
- Generalized HSD (G-HSD) — Beighton-positive GJH with musculoskeletal complications but not enough Feature A items, no qualifying family history, and no confirmed diagnosis of another EDS subtype.
- Peripheral HSD (P-HSD) — hypermobility limited to the hands and feet, with symptoms.
- Localized HSD (L-HSD) — hypermobility of a single joint or small joint group, with symptoms.
- Historical HSD (H-HSD) — self-reported historical GJH plus current musculoskeletal symptoms, when current Beighton is below threshold and historical GJH is established via the 5-point questionnaire.
The critical point for patients: HSD and hEDS are managed identically. Physical therapy, pacing, pain management, joint protection, and screening for the EDS-adjacent conditions (POTS, MCAS, GI dysmotility) are the same regardless of which label applies. The label is a research and communication tool, not a treatment-determining distinction. If a clinic refuses to treat HSD the way it treats hEDS, that clinic is out of date.
The Criteria Controversy
The 2017 criteria were designed to shrink the hEDS population in order to make gene-hunting feasible. That goal was reasonable. The side effect, which researchers openly acknowledge, is that many patients with clinically obvious EDS-type connective tissue involvement no longer qualify for the hEDS label.
Common ways patients "fail out" of the criteria:
- No family history. The affected parent died young, was never tested, or refuses to be evaluated. Without a confirmed first-degree relative, Feature B is unmet.
- Feature A short by one or two. Four of the twelve systemic signs instead of five — very common in younger patients whose striae, prolapse, or valve changes have not yet appeared.
- Beighton just under threshold in a now-stiffer adult, with incomplete historical documentation.
- Mixed phenotype that looks partly like classical EDS on skin exam but has a negative COL5 panel.
These patients are real, their symptoms are real, and their response to EDS-aware care is real. HSD is the correct label for most of them under the current rules, and HSD should open every door that hEDS opens — physical therapy referrals, genetics follow-up, cardiology screening, POTS and MCAS workups. In practice, some insurers and some clinicians treat HSD as a lesser diagnosis. That is a policy gap, not a medical one.
Self-Assessment at Home — The 5-Point Questionnaire
The Hakim and Grahame 5-point questionnaire is a validated screen you can do at your kitchen table. Two or more "yes" answers suggests generalized joint hypermobility:
- Can you now (or could you ever) place your hands flat on the floor without bending your knees?
- Can you now (or could you ever) bend your thumb to touch your forearm?
- As a child, did you amuse your friends by contorting your body into strange shapes or do the splits?
- As a child or teenager, did either of your shoulder or kneecap dislocate on more than one occasion?
- Do you consider yourself double-jointed?
Two or more "yes" answers is the cutoff. This is a screening tool, not a diagnosis — but if you score two or more and you have chronic unexplained pain, frequent sprains, fatigue, or dizziness on standing, the conversation with a hypermobility-aware physician is worth scheduling.
Finding a Knowledgeable Provider
EDS is under-taught in medical school. Most rheumatologists see one or two hEDS patients per year and may not know the 2017 criteria by heart. A smaller group has made hypermobility its focus. Practical ways to find one:
- The Ehlers-Danlos Society Healthcare Professionals Directory (ehlers-danlos.com) lists clinicians worldwide who self-identify as EDS-knowledgeable and have agreed to see EDS patients.
- Physiatry (PM&R) is often a better first stop than rheumatology. Hypermobility is a musculoskeletal-function problem, not an inflammatory-arthritis problem, and physiatrists think about joint mechanics and physical therapy dosing. Dr. Pradeep Chopra (Providence, RI) and physiatrists like Dr. Mark Driscoll are widely cited in the patient community.
- Clinical geneticists such as Dr. Clair Francomano (Indiana University / Harvey Institute tradition) have spent decades building EDS expertise and are among the researchers who wrote the criteria in the first place.
- Academic EDS clinics have emerged at several major centers (Johns Hopkins, Stanford, NIH, Cincinnati Children's for pediatrics). Waits are long — often 6 to 18 months — but these clinics are best equipped for complex multi-system cases.
- Local physical therapists with advanced hypermobility training fill the day-to-day gap. Ask whether they are familiar with the Muldowney protocol, the Levine protocol for POTS, or Chopra-style joint protection training.
Come to any first visit with a completed Beighton self-score, a timeline of dislocations and subluxations, a family tree sketch, and a list of associated symptoms (orthostatic intolerance, IBS-type GI complaints, skin findings, dental crowding). The encounter is short; your preparation is what makes it useful.
What the Genetic Test Gap Means
There is currently no genetic test that confirms hEDS. A negative EDS gene panel does not rule hEDS out, and a positive panel rules it in only for one of the other subtypes. Several active research threads are trying to close the gap:
- The HEDGE study (Hypermobile EDS Genetic Evaluation), coordinated by the Ehlers-Danlos Society, has enrolled thousands of strictly-criteria-meeting hEDS patients for whole-genome sequencing. Candidate variants are emerging but none has yet been validated for clinical use.
- COL5 subtype overlap. A subset of patients with COL5A1 variants present clinically more like hEDS than like textbook classical EDS — soft stretchy skin and hypermobility without the wide atrophic scars. These cases blur the line between the two subtypes and hint that the gene-phenotype map is not one-to-one.
- TNXB heterozygous variants. Biallelic TNXB loss causes a rare recessive EDS. Recent work suggests that heterozygous (single-copy) TNXB variants may contribute to a hEDS-like phenotype in some patients, although the penetrance and clinical significance are still being worked out.
- Mast cell and complement gene variants. Because hEDS so often travels with MCAS and dysautonomia, investigators are examining whether the unifying biology lies partly outside the collagen genes.
Until the gene is found, hEDS will remain a clinical diagnosis. That is not a failure of the patient; it is a current limitation of the science.
Classical EDS (cEDS) as a Contrast
Classical EDS is worth understanding briefly because it is the closest cousin of hEDS and the most common reason for a genetic exclusion panel. Under the 2017 criteria, a clinical diagnosis of cEDS requires the major criterion of skin hyperextensibility plus atrophic scars, together with either generalized joint hypermobility or at least three of nine minor criteria (easy bruising, soft doughy skin, fragile skin, molluscoid pseudotumors, subcutaneous spheroids, hernia, epicanthal folds, joint complications, and a positive first-degree family history). Confirmation requires a pathogenic variant in COL5A1 or COL5A2, and in a small fraction of cases a specific COL1A1 arginine-to-cysteine substitution.
The defining difference is the skin. cEDS skin splits easily with minor trauma and heals into wide, thin, papery scars — classically on the knees, elbows, shins, and forehead. hEDS skin may be soft, velvety, and mildly stretchy, but it does not usually produce the wide atrophic scarring. A careful skin exam can separate the two in most cases; a COL5 panel closes the question.
Cost Reality — Geneticist Visits and Panels
Pursuing a formal diagnosis has a price tag. Typical U.S. costs in 2026 dollars:
- Clinical geneticist consultation: roughly $300–$800 out of pocket if uncovered, less with insurance. Many academic centers bill a separate genetic-counselor fee on top. Telegenetics visits are increasingly available and generally cheaper.
- Targeted EDS gene panel (covering COL5A1, COL5A2, COL3A1, COL1A1, COL1A2, TNXB, and the rarer kyphoscoliotic and arthrochalasia genes): roughly $500–$1,500.
- Broader connective-tissue panel (adding Marfan and Loeys-Dietz genes, osteogenesis imperfecta expansion, cutis laxa genes, and others): roughly $1,500–$3,000.
- Whole-exome or whole-genome sequencing, if the panel is negative and clinical suspicion remains high: $1,000–$3,000 depending on the lab and whether trio testing (patient plus both parents) is included.
- Echocardiogram for aortic root measurement: $800–$2,500 billed, often fully covered by insurance when ordered for heritable connective tissue evaluation.
Insurance coverage has improved since 2017 but remains inconsistent. Labs such as Invitae, GeneDx, Prevention Genetics, and Blueprint Genetics offer EDS panels, and several will bill insurance directly or offer patient-assistance pricing that caps out-of-pocket costs around $100–$250 regardless of the list price. Ask your ordering clinician to request the patient-assistance quote in advance; it frequently saves hundreds to thousands of dollars.
A final note on expectations. Because hEDS has no gene yet, a negative panel does not rule hEDS out — it only rules out the subtypes it tests for. Patients sometimes walk into a genetics visit hoping for a definitive "yes, you have hEDS" result and walk out with a clinical diagnosis, a negative panel, and the same chronic symptoms. That is, oddly, a successful visit: the exclusion criterion of the 2017 rules has been met, and EDS-aware care can proceed.
Key Research Papers
- Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8–26.
- Castori M, Tinkle B, Levy H, Grahame R, Malfait F, Hakim A. A framework for the classification of joint hypermobility and related conditions. Am J Med Genet C Semin Med Genet. 2017;175(1):148–157.
- Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. (5-point questionnaire reference.) Am J Med Genet C Semin Med Genet. 2017.
- Tinkle B, Castori M, Berglund B, et al. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome Type III and Ehlers-Danlos syndrome hypermobility type): clinical description and natural history. Am J Med Genet C Semin Med Genet. 2017;175(1):48–69.
Live PubMed Searches
For further reading, the following PubMed topic searches return current peer-reviewed work on hypermobile EDS diagnosis, criteria, and associated conditions:
- Hypermobile EDS and the 2017 diagnostic criteria
- Beighton score and joint hypermobility assessment
- Hypermobility Spectrum Disorder (HSD) definition and management
- Genetic basis of hEDS and the HEDGE study
- TNXB variants and hypermobile EDS phenotype
- COL5A1 classical EDS and hEDS phenotype overlap
- Hakim 5-point questionnaire for joint hypermobility
- hEDS, POTS, and MCAS comorbidity
Connections
- Ehlers-Danlos Syndrome
- Classical and Vascular EDS
- Craniocervical Instability and AAI
- GI Involvement in EDS
- POTS, MCAS and the EDS Triad
- Pain Management in hEDS
- Pediatric EDS and Transition of Care
- Physical Therapy and Joint Protection
- Pregnancy and EDS
- POTS
- POTS Subtypes
- MCAS
- Fibromyalgia
- Chronic Pain
- Chronic Fatigue Syndrome
- Osteoporosis
- Collagen