Ehlers-Danlos Syndrome

Deep-Dive Articles
Ehlers-Danlos Syndrome (EDS) is not one condition but a family of at least thirteen inherited connective-tissue disorders, each with its own genetic signature, prognosis, and management priorities. The eight guides below drill into the parts that most often send patients looking for answers: the 2017 diagnostic framework for hypermobile EDS, the POTS/MCAS/EDS triad that defines daily life for many patients, the different stakes of classical and vascular EDS, practical pain control, a joint-protection physical-therapy approach, the surprisingly common GI involvement, pregnancy planning, and the pediatric-to-adult transition. Start with whichever matches where you are in your journey.
Hypermobile EDS & 2017 Criteria
The most common — and most contested — subtype. How the 2017 International Classification redefined hEDS, the three-part criteria (Beighton score, systemic features, family history/pain/instability), and why a negative genetic test doesn’t rule it out.
POTS, MCAS & the EDS Triad
The signature comorbid cluster: postural orthostatic tachycardia, mast cell activation, and hypermobile EDS. Why they co-occur, how to work up all three in parallel, and the treatment sequencing that gets patients functional again.
Classical & Vascular EDS
cEDS (COL5A1/COL5A2) with its atrophic scars and skin hyperextensibility, and vEDS (COL3A1) with its life-threatening arterial, bowel, and uterine rupture risk. Genetic testing, surveillance imaging, and the red flags that demand urgent cardiology referral.
Pain Management in hEDS
Why EDS pain is multi-source (joint subluxation, myofascial, neuropathic, central sensitization) and why opioids often fail. Practical layering: low-dose naltrexone, topical compounds, trigger-point work, bracing, and the integrative-medicine tactics that actually move the needle.
Physical Therapy & Joint Protection
The PT approach that works for hypermobile joints is the opposite of most PT: low-load, high-rep, closed-chain, proprioception-first. Muldowney, EDS ECHO protocols, bracing strategies, and the exercises that stabilize without grinding cartilage down.
GI Involvement in EDS
Gastroparesis, IBS, SIBO, rectal prolapse, hernias, and visceroptosis are all more common in EDS. The connective-tissue reasons behind it, which workup to push for, and the diet-plus-prokinetic protocols that restore upper-GI motility.
Pregnancy & EDS
Hypermobility-driven pelvic instability, preterm rupture of membranes, precipitous labor, postpartum flares, and — in vEDS — catastrophic rupture risk. Pre-conception counseling, high-risk OB referral, and which subtypes contraindicate pregnancy altogether.
Pediatric EDS & Transition of Care
Kids with EDS are often labeled “clumsy,” “growing pains,” or “anxious” for years before diagnosis. Developmental red flags, school accommodations (IEP/504), pacing strategies, and the handoff from pediatrics to adult rheumatology/genetics that too often breaks down.
Craniocervical Instability (CCI) & AAI
Upright MRI, clivo-axial angle, and the controversy around C0-C2 fusion surgery.
1. Overview
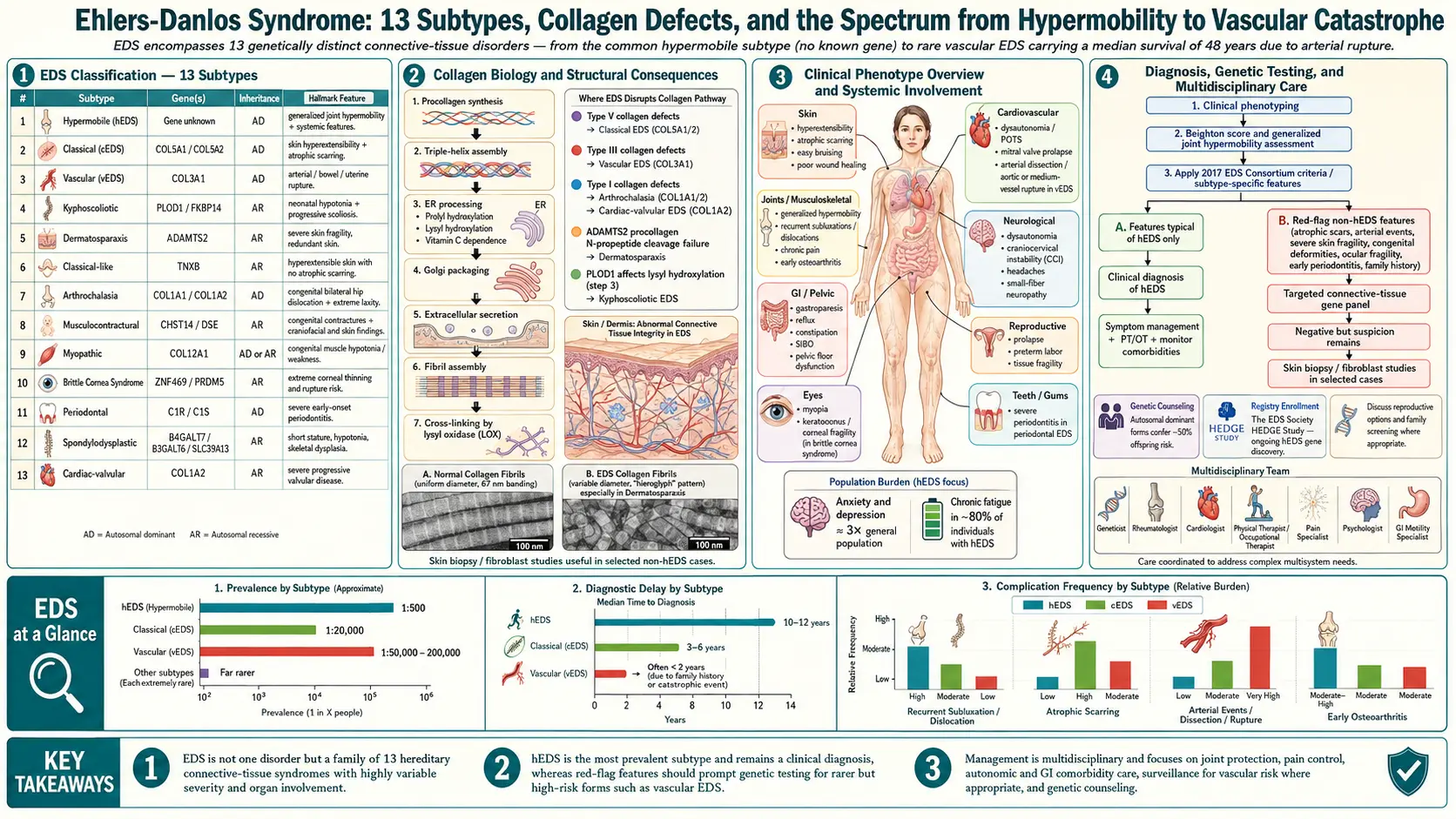
Ehlers-Danlos syndrome (EDS) is not a single disease but a group of approximately 13 heritable connective-tissue disorders caused by defects in the synthesis, processing, or assembly of collagen — the protein scaffolding that gives skin, joints, blood vessels, and hollow organs their structural integrity. Because collagen is everywhere in the body, EDS is a multi-system disorder. The shared clinical thread across subtypes is some combination of joint hypermobility, skin hyperextensibility, and tissue fragility, but the severity and organ involvement vary dramatically — from a person with mildly bendy fingers and bruising to a patient at risk of sudden arterial rupture.
The current reference framework is the 2017 International Classification of the Ehlers-Danlos Syndromes, which recognizes 13 distinct subtypes defined by clinical criteria, mode of inheritance, and, for twelve of them, a confirmed genetic defect. The thirteenth — hypermobile EDS (hEDS) — is the most common but has no identified causative gene, which means its diagnosis remains clinical and is sometimes contested. Most other subtypes can now be confirmed with next-generation sequencing panels covering COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, ADAMTS2, PLOD1, FKBP14, B4GALT7, and other collagen-related genes.
EDS is under-diagnosed. The average patient sees 5 to 20 different clinicians over 10 to 20 years before receiving a correct label, and the delay is longer in women, in whom symptoms are often dismissed as anxiety, deconditioning, or fibromyalgia. Understanding the disease — and having a short, clear way to explain it to new providers — is one of the most protective things a patient can do.
2. The 2017 International Classification — 13 Subtypes
The 2017 classification, published by Malfait and colleagues, organized EDS into thirteen subtypes. The major ones you are most likely to encounter are:
- Hypermobile EDS (hEDS) — by far the most common subtype; autosomal dominant; no identified gene; diagnosed purely by the 2017 clinical criteria (Beighton score, a checklist of systemic connective-tissue features, plus family history, musculoskeletal complications, or pain).
- Classical EDS (cEDS) — caused by pathogenic variants in COL5A1 or COL5A2 (rarely COL1A1); autosomal dominant; characterized by marked skin hyperextensibility, atrophic “cigarette-paper” scars, and generalized joint hypermobility.
- Vascular EDS (vEDS) — caused by variants in COL3A1 (rarely COL1A1); autosomal dominant; the dangerous subtype — arterial dissection and rupture, bowel perforation, and uterine rupture can occur without warning. Average life expectancy historically ~50 years; modern surveillance and celiprolol may extend it.
- Kyphoscoliotic EDS (kEDS) — autosomal recessive; PLOD1 or FKBP14; congenital hypotonia, progressive scoliosis, and scleral fragility.
- Arthrochalasia EDS (aEDS) — COL1A1 or COL1A2; severe joint hypermobility with bilateral congenital hip dislocation.
- Dermatosparaxis EDS (dEDS) — ADAMTS2; extremely fragile, sagging skin.
- Classical-like EDS (clEDS) — TNXB; a recessive mimic of cEDS.
- Cardiac-valvular EDS (cvEDS) — COL1A2; progressive cardiac valvular disease.
- Spondylodysplastic EDS (spEDS) — B4GALT7, B3GALT6, SLC39A13.
- Musculocontractural EDS (mcEDS) — CHST14, DSE.
- Myopathic EDS (mEDS) — COL12A1; congenital muscle weakness with contractures.
- Periodontal EDS (pEDS) — C1R, C1S; early severe periodontitis and tooth loss.
- Brittle cornea syndrome (BCS) — ZNF469, PRDM5; thin cornea and risk of rupture.
Newer types proposed since 2017 include a hEDS-related disorder linked to AEBP1 and other candidate genes under active study.
3. Prevalence and Under-Diagnosis
Collectively, all EDS subtypes together are estimated to affect roughly 1 in 5,000 people, though many experts believe the true figure is higher because most cases go unrecognized. Hypermobile EDS is by far the most common subtype, with estimates ranging from 1 in 3,000 to 1 in 5,000 — and some clinical researchers, including Rodney Grahame, argue that the prevalence of the broader hypermobility spectrum disorders (HSD) + hEDS may be as high as 1 in 600.
The rare subtypes are genuinely rare: vascular EDS is estimated at 1 in 50,000 to 1 in 200,000; classical EDS at roughly 1 in 20,000 to 1 in 40,000; kyphoscoliotic and other recessive subtypes are rarer still.
Historically EDS has been dramatically under-diagnosed, especially in women, who make up an estimated 75-90% of diagnosed hEDS cases but are routinely misdiagnosed with fibromyalgia, chronic fatigue, anxiety, or somatization first. The typical diagnostic odyssey — 5 to 20 providers, 10 to 20 years, multiple wrong labels — is itself a source of trauma that the EDS community calls “medical gaslighting.” The 2017 criteria and the emergence of genetics-literate rheumatologists and geneticists have begun to shorten this delay, but access remains a bottleneck — there are only a few hundred EDS-experienced clinicians in the United States for millions of potential patients.
4. Core Clinical Features
Joint Hypermobility and the Beighton Score
The universal finding in most EDS subtypes is generalized joint hypermobility (GJH), measured using the Beighton 9-point score:
- Passive dorsiflexion of the 5th finger beyond 90° (1 point each side)
- Passive apposition of the thumb to the flexor forearm (1 point each side)
- Hyperextension of the elbow beyond 10° (1 point each side)
- Hyperextension of the knee beyond 10° (1 point each side)
- Forward flexion of the trunk with palms flat on the floor, knees straight (1 point)
A score of ≥6/9 in pre-pubertal children, ≥5/9 in adults, and ≥4/9 in those over 50 is considered generalized hypermobility. The 5-Point Questionnaire can capture historical hypermobility in people whose joints have stiffened with age.
Skin Findings
- Hyperextensibility — skin can be pulled farther than normal (especially obvious in cEDS).
- Atrophic scarring — thin, widened, “cigarette-paper” scars, particularly over the knees and shins.
- Easy bruising — frequent bruises from minor trauma; piezogenic heel papules.
- Soft, velvety, translucent skin — visible veins through the chest and abdomen, especially in vEDS.
Tissue Fragility
Poor wound healing, recurrent hernias, rectal or uterine prolapse, cervical insufficiency in pregnancy, and (in vEDS) spontaneous rupture of arteries, bowel, or uterus.
Pain and Fatigue
Chronic widespread pain is nearly universal in hEDS and cEDS. Sources include recurrent subluxations, myofascial pain, early-onset osteoarthritis, neuropathic pain, headaches (migraine, cervicogenic, craniocervical instability), and central sensitization. Fatigue is often the most disabling symptom — frequently reaching ME/CFS severity in hEDS.
5. The hEDS Comorbidity Cluster
Hypermobile EDS rarely travels alone. The recognized comorbidity profile includes:
- The POTS / MCAS / hEDS triad — postural orthostatic tachycardia syndrome affects ~40-70% of hEDS patients; mast cell activation syndrome (flushing, food reactivity, hives, anaphylaxis) is similarly common. All three share connective-tissue and autonomic threads; see the POTS/MCAS/EDS triad deep dive.
- Gastrointestinal dysmotility — gastroparesis, IBS (often IBS-C), SIBO, GERD, visceroptosis, rectal prolapse.
- Autonomic dysfunction — beyond POTS, including temperature dysregulation, sleep disruption, and sweating changes.
- Chronic pain syndromes — fibromyalgia overlap, small-fiber neuropathy (present in ~30% on skin biopsy), complex regional pain syndrome.
- Mental health — anxiety and depression at significantly higher rates than controls; this is biological (interoception, autonomic arousal), not psychogenic.
- Pelvic-floor dysfunction — incontinence, prolapse, dyspareunia.
- Craniocervical instability and Chiari-like headaches — in a subset of severe hEDS; upright MRI may be needed.
- Dental crowding, high-arched palate, TMJ dysfunction — common and often an early clue in children.
- Endometriosis and heavy menstrual bleeding — both over-represented.
6. Diagnostic Pathway
A reasonable diagnostic sequence, in order:
- Thorough personal and family history. Ask about: childhood clumsiness, subluxations, growing pains, bruising, poor scarring, stretchy skin, dental work history, GI symptoms, fainting, food reactions, and family members with similar features or sudden cardiac/vascular deaths.
- Physical exam. Beighton score, skin assessment (stretch, scars, softness), atrophic scarring, palate, spine, hindfoot alignment, hernia scars, and piezogenic papules.
- Apply the 2017 criteria. For hEDS: all three criteria must be met — (1) generalized joint hypermobility, (2) two or more systemic manifestations / positive family history / musculoskeletal complications, (3) exclusion of other diagnoses.
- Referral to medical genetics or EDS-experienced rheumatology. Genetic testing with a multi-gene connective-tissue panel is appropriate when any red flags for a non-hypermobile subtype are present. A negative panel does not rule out hEDS because no gene has been identified.
- Red-flag screening for vEDS. Family history of arterial or hollow-organ rupture, translucent skin with visible veins, acrogeria (aged-looking hands), easy bruising, thin lips and pinched nose, or arterial dissection at a young age — any of these demand urgent COL3A1 testing and referral to a vascular specialist, because management changes immediately.
7. Vascular EDS — Red Flags That Cannot Wait
Vascular EDS deserves its own section because a missed diagnosis can be fatal. Call it out for urgent workup when you see:
- Family history of arterial rupture or dissection, bowel rupture, uterine rupture, or sudden unexplained death before age 50.
- Thin, translucent skin with a visible venous pattern on the upper chest.
- Acrogeria — prematurely aged appearance of the hands.
- Characteristic facies — thin lips, small chin, pinched nose, prominent eyes.
- Easy, extensive bruising out of proportion to trauma.
- Early varicose veins, carotid-cavernous fistula, pneumothorax, gum recession.
- Arterial dissection, aneurysm, or rupture at a young age — even a single event in someone under 40.
Confirmed vEDS requires: COL3A1 molecular confirmation, surveillance imaging (CT or MR angiography of chest, abdomen, pelvis, head, and neck at baseline and then on interval), consideration of celiprolol (a beta-1 antagonist shown in the BBEST trial to reduce vascular events), avoidance of elective arterial catheterization and contact sports, and a clear emergency plan that every hospital in the patient’s region knows about. Pregnancy in vEDS carries a 5-10% maternal mortality risk per pregnancy and requires specialist counseling.
8. Treatment Framework
There is no cure for EDS. Management is multidisciplinary, patient-led, and lifelong. A well-assembled team typically includes:
- A primary care clinician who is willing to learn and coordinate.
- Medical genetics for diagnosis, family counseling, and surveillance recommendations.
- Rheumatology for joint care and comorbid autoimmune workup.
- Cardiology — mandatory in vEDS (serial aortic imaging) and recommended in hEDS for baseline echocardiogram and POTS workup.
- Physical therapy with an EDS-aware clinician using low-load, proprioception-first protocols (Muldowney, EDS ECHO).
- Pain management favoring multimodal non-opioid strategies — low-dose naltrexone, topical compounds, medical cannabis where legal, trigger-point injections.
- Gastroenterology for dysmotility, SIBO, and prolapse.
- Allergy/immunology for MCAS workup.
- Autonomic neurology or cardiology for POTS (tilt table, fluids, compression garments, midodrine, ivabradine, beta-blockers).
- Mental-health support — ideally a therapist experienced with chronic illness and trauma.
- Pelvic-floor PT, dentistry, ophthalmology (keratoconus surveillance in kEDS/BCS), and, in selected cases, neurosurgery (for craniocervical instability).
Day-to-day self-management centers on pacing, joint protection, hydration and salt (for orthostatic symptoms), a regular low-load exercise program, and avoidance of the hyperextension positions that trigger subluxations. Bracing (ring splints for fingers, SI belts, cervical collars during flares) is a frequent underused tool.
9. Registries, Organizations, and Research
- The Ehlers-Danlos Society — the largest international EDS advocacy and research organization; runs the EDS ECHO professional-education program and coordinates multinational research.
- International Consortium on EDS and HSD — the body that produced the 2017 classification and continues to refine diagnostic criteria.
- The vEDS Collaborative — patient-and-researcher network focused specifically on vascular EDS, with a global registry and natural-history studies.
- Annals Clinical and Translational Neurology / AJMG-C special issues — the original 2017 framework papers are the foundational reading.
- Hereditary Disorders of Connective Tissue (HDCT) Program — multiple academic medical centers (Hopkins, Cincinnati Children’s, NIH Undiagnosed Diseases Program) maintain specialty clinics.
Active research areas include identifying the hEDS gene(s), refining diagnostic biomarkers (skin biopsy for fibrillin/collagen ratios, small-fiber neuropathy), clinical trials of celiprolol and emerging agents in vEDS, and mechanistic work on why autonomic and mast-cell dysfunction co-aggregate with hypermobility.
10. Your Deep-Dive Roadmap
The eight deep-dive articles linked at the top of this page expand the sections above into practical patient guides:
- Hypermobile EDS & 2017 Criteria — the detailed checklist, common misapplications, and how to advocate for the diagnosis.
- POTS, MCAS & the EDS Triad — the mechanistic links, parallel workup, and treatment sequencing.
- Classical & Vascular EDS — genetic testing, surveillance, and the emergency-plan template for vEDS.
- Pain Management in hEDS — the layered non-opioid approach that most rheumatology offices never write down.
- Physical Therapy & Joint Protection — the EDS-specific PT protocol and bracing strategy.
- GI Involvement in EDS — gastroparesis, SIBO, prolapse, and what to push for.
- Pregnancy & EDS — pre-conception planning through postpartum flares.
- Pediatric EDS & Transition of Care — childhood clues, school accommodations, and the handoff to adult care.
Read whichever card matches where you are today. EDS care is iterative — you do not have to solve everything at once.
Table of Contents
- Deep-Dive Articles
- Overview
- The 2017 International Classification
- Prevalence and Under-Diagnosis
- Core Clinical Features
- The hEDS Comorbidity Cluster
- Diagnostic Pathway
- Vascular EDS Red Flags
- Treatment Framework
- Registries, Organizations, Research
- Your Deep-Dive Roadmap
- Key Research Papers
- Connections
- Featured Videos
Key Research Papers
Foundational primary references with live DOI links, followed by curated PubMed topic searches that stay current as new studies are indexed.
- Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
- Tinkle B, Castori M, Berglund B, et al. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome Type III and Ehlers-Danlos syndrome hypermobility type): Clinical description and natural history. Am J Med Genet C Semin Med Genet. 2017;175(1):48-69.
- Castori M, Tinkle B, Levy H, et al. A framework for the classification of joint hypermobility and related conditions. Am J Med Genet C Semin Med Genet. 2017;175(1):148-157.
- Tinkle B, Levy H. Symptomatic joint hypermobility: The hypermobile type of Ehlers-Danlos syndrome and the hypermobility spectrum disorders. Clinical approach, Am J Med Genet C. 2017.
- Pepin MG, Schwarze U, Rao VR, et al. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med. 2014;16(12):881-888.
Curated PubMed topic searches on Ehlers-Danlos Syndrome:
- PubMed topic search: Ehlers-Danlos syndrome review
- PubMed topic search: hypermobile EDS 2017 criteria
- PubMed topic search: vascular EDS COL3A1
- PubMed topic search: classical EDS COL5A1
- PubMed topic search: POTS and EDS
- PubMed topic search: MCAS and EDS
- PubMed topic search: Beighton score hypermobility
- PubMed topic search: EDS gastroparesis dysmotility
- PubMed topic search: celiprolol vascular EDS (BBEST trial)
- PubMed topic search: pregnancy and EDS outcomes
- PubMed topic search: small fiber neuropathy in EDS
- PubMed topic search: craniocervical instability EDS
Connections
- Hypermobile EDS and 2017 Diagnostic Criteria
- Classical and Vascular EDS
- Craniocervical Instability and AAI
- GI Involvement in EDS
- POTS, MCAS and the EDS Triad
- Pain Management in hEDS
- Pediatric EDS and Transition of Care
- Physical Therapy and Joint Protection
- Pregnancy and EDS
- POTS
- POTS-MCAS-EDS Triad
- MCAS
- Fibromyalgia
- Chronic Fatigue Syndrome
- Chronic Pain
- Migraine
- Osteoporosis
- Collagen