Classical and Vascular EDS
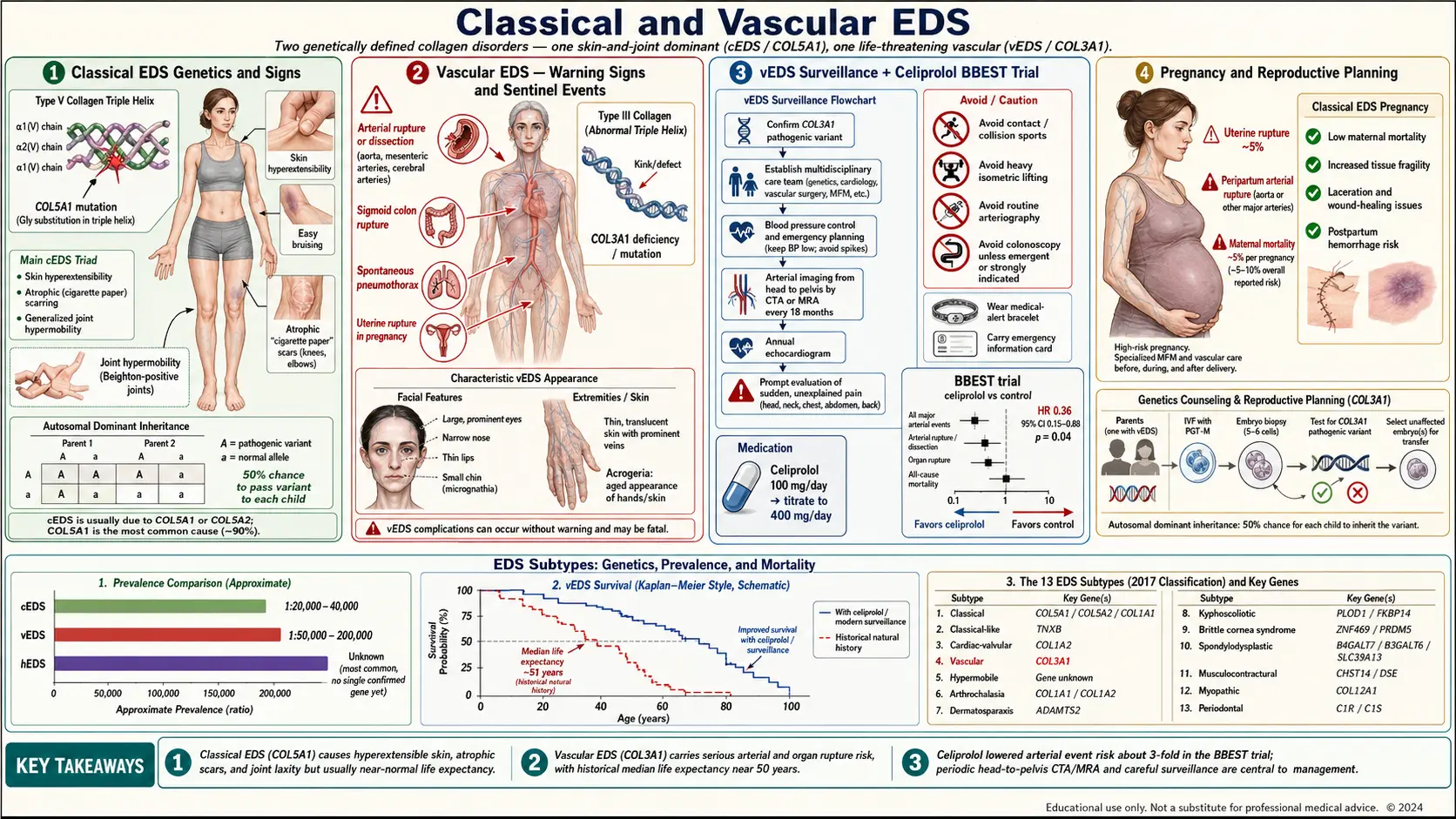
Table of Contents
- Why Classical and Vascular EDS Are Grouped Here
- Classical EDS — Genetics and How Common
- Classical EDS — What It Looks and Feels Like
- Classical EDS — The 2017 Diagnostic Criteria
- Classical EDS — Day-to-Day Management
- Vascular EDS — Genetics and How Common
- Vascular EDS — The Warning Signs
- Vascular EDS — Sentinel Events and Life Expectancy
- Vascular EDS — Urgent Workup and Surveillance
- Vascular EDS — Celiprolol and the BBEST Trial
- Vascular EDS — Lifestyle, Surgery, Emergency Prep
- Vascular EDS and Pregnancy
- Other Rarer EDS Subtypes in Brief
- Genetic Counseling and Reproductive Planning
- Key Research Papers
- Connections
- Featured Videos
Why Classical and Vascular EDS Are Grouped Here
Of the thirteen subtypes recognized in the 2017 international EDS classification, classical EDS (cEDS) and vascular EDS (vEDS) are the two that almost every clinician has heard of, and the two where a specific collagen gene mutation can be confirmed in a blood test. They also sit at opposite ends of the risk spectrum. Classical EDS is the "textbook" connective-tissue disorder — stretchy skin, wide scars, wobbly joints — and while it complicates life, it rarely shortens it. Vascular EDS is the form that can kill you without warning. Understanding which one is on the table — or whether a third, less alarming subtype fits better — changes everything about surveillance, pregnancy planning, emergency care, and how worried a family should be.
This article walks through both in patient-friendly language, explains how they differ from hypermobile EDS (the form most EDS patients have), and gives a brief map of the other rarer subtypes so you know where your diagnosis fits on the larger landscape.
Classical EDS — Genetics and How Common
Classical EDS is caused in more than 90% of cases by a mutation in COL5A1 or COL5A2, the two genes that code for type V collagen. Type V is a minor collagen by volume but a major regulator: it acts like a quality-control supervisor for type I collagen fibril assembly in skin, tendon, and blood vessel walls. Lose half the working copies of COL5A1 (the usual mechanism — haploinsufficiency) and the collagen bundles in your skin end up disorganized, too large, and mechanically weak.
Inheritance is autosomal dominant. One copy of the mutation is enough to cause disease, each child of an affected parent has a 50% chance of inheriting it, and men and women are affected equally. A small minority of cases (a few percent) carry a specific mutation in COL1A1 at position c.934C>T (p.Arg312Cys) that produces a classical-EDS-like phenotype, sometimes with extra vascular risk.
Estimated prevalence is roughly 1 in 20,000 to 1 in 50,000. That puts it well ahead of vascular EDS for frequency but well behind hypermobile EDS, which is not rare at all and may affect 1 in 500 to 1 in 5,000 depending on how it is counted.
Classical EDS — What It Looks and Feels Like
The three findings that define classical EDS in the clinic are skin hyperextensibility, atrophic scarring, and generalized joint hypermobility. Each deserves a closer look.
Skin hyperextensibility. This is tested by the pinch-and-pull maneuver: a clinician grabs a fold of skin on the volar forearm (palm side) away from joints and lifts it. Classical-EDS skin stretches dramatically — typically more than 1.5 cm — and then snaps back smartly when released. It does not hang loose like cutis laxa; it recoils. The skin also feels distinctly soft and doughy, a texture most patients and their parents recognize without prompting.
Atrophic "cigarette-paper" scars. When classical-EDS skin is cut, punctured, or scraped, it heals with a wide, thin, papery scar that often darkens over time and appears to float slightly below the surrounding skin. Knees, shins, elbows, chin, and forehead are the classic sites because these are the places children fall and adults bang into things. By adulthood many patients have dozens of these scars. They are the single most specific sign for the subtype.
Generalized joint hypermobility. Measured by the Beighton score (a 9-point examination of thumb-to-forearm, fifth-finger extension, elbow hyperextension, knee hyperextension, and palms-to-floor). Most classical-EDS patients score 5 or higher in childhood, though flexibility tends to decrease with age.
Other common findings: easy bruising disproportionate to trauma, molluscoid pseudotumors (small fleshy lumps that form at pressure points like the heels and over scars), spheroid subcutaneous masses (pea-sized mobile lumps you can roll under the skin, often on shins and forearms — these are calcified fat cysts), poor wound healing, and a tendency toward hernias, rectal prolapse in childhood, and cervical insufficiency in pregnancy.
Classical EDS — The 2017 Diagnostic Criteria
The international consortium led by Fransiska Malfait published updated criteria in 2017 (American Journal of Medical Genetics, DOI below). For classical EDS they define:
Major criteria (both must be present):
- Skin hyperextensibility and atrophic scarring
- Generalized joint hypermobility (Beighton score ≥ 5 in adults, ≥ 6 in children and adolescents, or a history of it)
Minor criteria (contribute to the clinical picture):
- Easy bruising
- Soft, doughy skin
- Skin fragility (or traumatic splitting)
- Molluscoid pseudotumors
- Subcutaneous spheroids
- Hernias
- Epicanthic folds
- Complications of joint hypermobility (sprains, subluxations, pes planus)
- Family history of a first-degree relative meeting criteria
A clinical diagnosis requires both major criteria plus at least three minor criteria. A definitive diagnosis requires genetic confirmation — a pathogenic variant in COL5A1, COL5A2, or (rarely) the specific COL1A1 Arg312Cys substitution. Modern panels run all three for a few hundred dollars and are covered by most insurance when clinical criteria are met.
Classical EDS — Day-to-Day Management
There is no disease-modifying drug for classical EDS. Management is about protecting tissue that does not heal well and screening for the small but real complications.
Wound care. Any cut in classical-EDS skin should be taken seriously. Ask your surgeon or emergency physician for:
- Longer suture retention — typically twice as long as standard (14 days on the face instead of 5–7; 21 days on the trunk instead of 10–14).
- Double-layer closure when possible — a deep absorbable suture layer plus a surface layer takes tension off the skin edge.
- Wound reinforcement with adhesive strips (Steri-Strips) for weeks after sutures come out.
- Longer splinting of ligament and tendon injuries — typical healing times are about 50% longer than textbook.
Physical therapy and joint protection. A PT who knows connective-tissue disease can design a low-impact strengthening program that stabilizes joints without stretching already-lax ligaments. Isometric work, closed-chain exercises, bracing of the most unstable joints (wrists, ankles, patellae), and swimming are cornerstones. Contact sports and heavy weightlifting are generally discouraged in adolescence, though this is individualized.
Cardiac screening. Classical EDS carries a small but measurable risk of aortic root dilation — lower than vascular EDS but not zero. Most specialists recommend a baseline echocardiogram at diagnosis and repeat imaging every 3–5 years if the root is normal, more often if it is enlarged. Mitral valve prolapse is also more common than in the general population.
Dental and oral care. Gingival fragility is common; soft-bristle brushes, careful flossing, and alerting dentists before cleanings (which can cause bruising) reduce trouble.
Life expectancy. Normal. Classical EDS is a quality-of-life disease, not a life-shortening one.
Vascular EDS — Genetics and How Common
Vascular EDS is caused in more than 95% of confirmed cases by a mutation in COL3A1, the gene for type III collagen. Type III is the collagen that reinforces the walls of medium and large arteries, the bowel, and the uterus — precisely the tissues that rupture in this disease. A small number of cases are caused by rare variants in COL1A1 or in the gene for type I collagen processing enzymes.
Inheritance is again autosomal dominant, though roughly half of cases are de novo — the affected person is the first in their family, with no parental history. Estimated prevalence is 1 in 50,000 to 1 in 200,000, though many experts suspect it is under-diagnosed because sudden deaths from unexplained dissection or bowel rupture are often not worked up genetically.
Vascular EDS — The Warning Signs
Vascular EDS does not look like classical EDS. The skin is not especially hyperextensible; instead it is thin and translucent, so much so that a web of veins is clearly visible on the chest, breasts, and abdomen in adults who are not thin. The face has a distinctive appearance called acrogeria: a thin narrow face, prominent cheekbones, thin nose, thin lips, hollowed cheeks, large-looking eyes that appear to protrude, and earlobes that are small or completely absent. Hands and feet look older than the person's age.
Joint hypermobility, if present, is usually limited to the small joints of the fingers and toes, not the large-joint generalized hypermobility of classical or hypermobile EDS. Many vEDS patients do not feel unusually flexible at all.
The feature that is usually dramatic is bruising. Spontaneous or very-light-trauma bruises — sometimes large, sometimes appearing in sheets — are often the first thing a parent or partner notices. Children with vEDS are sometimes investigated for child abuse or clotting disorders before the real diagnosis is made. Varicose veins in young people, premature aging of the hands, and a history of pneumothorax (collapsed lung) also show up repeatedly.
Vascular EDS — Sentinel Events and Life Expectancy
The reason vEDS is feared is that the first serious presentation is often a sentinel event — a sudden, severe, sometimes fatal rupture of an artery or hollow organ. The three classic events are:
- Arterial dissection or rupture. Any medium or large artery is at risk. Aorta, carotid, vertebral, renal, mesenteric, iliac, splenic — all have been reported. Presentation is typically sudden severe pain (chest, abdomen, flank, or neck), stroke-like symptoms, or collapse.
- Bowel rupture. The sigmoid colon is the classic site. Presents as sudden severe abdominal pain and peritonitis in a young person with no prior GI history.
- Uterine rupture during pregnancy or delivery — discussed below.
The landmark 2014 natural-history study by Melanie Pepin and Peter Byers at the University of Washington (Genetics in Medicine, DOI below) followed 1,231 confirmed COL3A1-positive patients and found:
- Median age at first major complication: 29 years.
- Roughly 80% had a major vascular event by age 40.
- Historical median survival: about 48 years, with most deaths from arterial rupture.
These are grim numbers, but they reflect a cohort diagnosed largely after a sentinel event. Earlier genetic diagnosis, aggressive surveillance, and celiprolol are shifting the curve. Patients diagnosed proactively — often because a relative had an event — are doing substantially better than the historical average.
Vascular EDS — Urgent Workup and Surveillance
If vEDS is suspected clinically, the workup moves fast. A reasonable protocol:
- Genetic testing. A COL3A1 sequencing panel (usually a broader connective-tissue panel that also covers COL1A1, COL1A2, COL5A1, COL5A2, and the aortopathy genes) is ordered right away. Results typically take 2–4 weeks.
- Baseline whole-body magnetic resonance angiography (MRA) from the circle of Willis through the iliac arteries. MRA is preferred over CT angiography because it avoids iodinated contrast (which can cause dissection at the catheter site if arterial access is used) and avoids radiation in patients who will need repeated imaging. Repeat every 12–24 months for life, or sooner if symptoms change.
- Transthoracic echocardiogram at baseline to measure aortic root diameter and check valve function; repeat annually or biennially.
- Baseline head and neck MRA to screen for intracranial aneurysms.
- Referral to a vascular surgeon who has treated vEDS before — emergency surgery on vEDS tissue is technically very different from standard vascular surgery and benefits enormously from experience.
Vascular EDS — Celiprolol and the BBEST Trial
For decades there was no drug proven to change vEDS outcomes. That changed with the BBEST trial (Beta-Blockers in Ehlers-Danlos Syndrome Treatment), published by Ong, Ardant, and colleagues in The Lancet in 2010. The trial randomized 53 patients with clinically diagnosed vEDS (33 with confirmed COL3A1 mutations) to celiprolol — a cardioselective beta-blocker with partial beta-2 agonist activity — versus no treatment, and followed them for a mean of five years.
Celiprolol reduced arterial events (dissection or rupture) by roughly 36% relative risk — enough of a benefit that the trial was stopped early for efficacy. Celiprolol became the first and still the only vEDS-specific drug endorsed by international guidelines.
Typical dosing starts at 100 mg twice daily and titrates up to 200 mg twice daily (total 400 mg/day) as blood pressure and heart rate tolerate. The goal is not aggressive blood-pressure lowering but a steady reduction in arterial wall stress. Celiprolol is not sold in the United States and has to be imported — a frustrating logistical fact that vEDS clinics help patients navigate. Alternative beta-blockers (atenolol, metoprolol, bisoprolol) are sometimes substituted when celiprolol is unobtainable, though the evidence for those is weaker. Aggressive blood-pressure control generally, avoiding hypertensive spikes, is a goal for everyone with vEDS — see the hypertension page for general BP management strategies.
Vascular EDS — Lifestyle, Surgery, Emergency Prep
Lifestyle modifications matter because tissue fragility compounds every risk.
- No contact sports. Football, rugby, hockey, boxing, martial arts, and most collision activities are out. Swimming, walking, light cycling, yoga (careful with extreme ranges), and low-weight strength training are generally safe.
- No heavy lifting or Valsalva straining. Anything that spikes intra-abdominal or intra-thoracic pressure (heavy deadlifts, aggressive weight training, straining on the toilet) is discouraged. Treat constipation aggressively with osmotic laxatives and fiber.
- Avoid NSAIDs. Ibuprofen, naproxen, aspirin, and other NSAIDs increase bleeding risk into already-fragile tissue. Acetaminophen is the default analgesic.
- Avoid arterial catheterization when possible. Elective coronary angiography, arterial lines, and cardiac catheterization carry real dissection risk. Non-invasive imaging is preferred. When catheterization is genuinely necessary, it should be done at a center experienced with vEDS.
- Medical ID bracelet and wallet card. Every vEDS patient should carry clear documentation: the diagnosis, COL3A1 mutation, medications (celiprolol), and a one-line instruction to emergency teams: "Avoid arterial catheterization; tissue is fragile; call genetics/vascular surgery."
- Surgery caution. Any surgery on vEDS tissue — whether elective or emergent — is technically harder. Sutures tear through, vessels do not hold clips well, and hemostasis is harder to achieve. Surgeons who know this operate more slowly, use pledgeted sutures, avoid aggressive retraction, and plan for longer recoveries.
Vascular EDS and Pregnancy
Pregnancy is the highest-risk time for a vEDS patient. Historical mortality figures cite roughly 12–15% maternal mortality per pregnancy, driven by arterial rupture, uterine rupture, and postpartum hemorrhage. Modern care at experienced centers brings this down substantially but never to baseline.
Decisions about whether to attempt pregnancy are deeply personal and deserve detailed counseling with a maternal-fetal-medicine team, a vascular surgeon, a geneticist, and the patient's partner. When pregnancy is undertaken, delivery is typically planned at a tertiary center with vascular surgery on standby, and the mode of delivery (scheduled cesarean versus vaginal) is individualized. Preimplantation genetic diagnosis is available for COL3A1 (see counseling section below). See also the Pregnancy and EDS article for the broader EDS-and-pregnancy picture.
Other Rarer EDS Subtypes in Brief
The 2017 classification recognizes thirteen EDS subtypes. Beyond hypermobile, classical, and vascular, the others are rare but worth naming so patients know whether to seek out a specialist:
- Kyphoscoliotic EDS — PLOD1 or FKBP14 mutations; severe scoliosis from birth, muscle hypotonia, globe fragility.
- Arthrochalasia EDS — COL1A1 or COL1A2; congenital bilateral hip dislocation and extreme joint laxity.
- Dermatosparaxis EDS — ADAMTS2; extremely fragile, saggy, redundant skin.
- Classical-like EDS — TNXB (tenascin-X deficiency); looks like classical EDS but with normal scars.
- Spondylodysplastic EDS — short stature, bowed limbs.
- Musculocontractural EDS — CHST14 or DSE; contractures, distinctive face.
- Myopathic EDS — COL12A1; muscle weakness plus hypermobility.
- Cardiac-valvular EDS — COL1A2; progressive severe valve disease.
- Periodontal EDS — C1R or C1S; early aggressive periodontal disease.
- Brittle cornea syndrome — sometimes grouped with EDS; ZNF469 or PRDM5.
If any of these labels fit your clinical picture better than hypermobile EDS, ask your geneticist to broaden the sequencing panel. Many of them have specific surveillance recommendations that are missed if the diagnosis is left at "EDS, unspecified."
Genetic Counseling and Reproductive Planning
Any patient with confirmed or strongly suspected classical or vascular EDS benefits from at least one session with a board-certified genetic counselor. The counselor can:
- Explain inheritance risk to children (50% for both cEDS and vEDS in dominant forms).
- Arrange cascade testing for at-risk relatives — especially important in vEDS, where a diagnosis can save a sibling or parent's life.
- Discuss preimplantation genetic diagnosis (PGD), available for COL3A1 and other single-gene EDS mutations. PGD involves IVF with embryo biopsy and selection of embryos that do not carry the mutation. Costs range from $15,000 to $30,000 per cycle and are sometimes partially covered by insurance when a severe dominant condition is involved.
- Discuss prenatal diagnosis (chorionic villus sampling at 10–13 weeks, amniocentesis at 15–20 weeks) for families who conceive naturally.
- Address the emotional side — grief, guilt, and the difficulty of telling family members about a dominant condition.
For vEDS, early diagnosis of relatives is not optional in the same way hEDS diagnosis sometimes feels optional. A 25-year-old second cousin who turns out to carry COL3A1 can be started on celiprolol and surveillance imaging before the first dissection — the kind of intervention that changes the rest of a life.
Key Research Papers
- Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017.
- Bowen JM, Sobey GJ, Burrows NP, et al. Ehlers-Danlos syndrome, classical type. Am J Med Genet C Semin Med Genet. 2017. (Byers and colleagues, classical-EDS review)
- Pepin MG, Schwarze U, Rice KM, et al. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med. 2014.
- Ong KT, Perdu J, De Backer J, et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial (BBEST). Lancet. 2010.
Live PubMed Searches
For further reading, the following PubMed topic searches return current peer-reviewed work on classical and vascular EDS:
- Classical EDS and COL5A1/COL5A2 mutations
- Vascular EDS and COL3A1 mutations
- Celiprolol in vascular EDS
- Aortic root dilation in EDS
- Vascular EDS and pregnancy outcomes
- Wound healing and surgical considerations in EDS
- 2017 international EDS classification
- Preimplantation genetic diagnosis for COL3A1
Connections
- Ehlers-Danlos Syndrome
- Hypermobile EDS and 2017 Diagnostic Criteria
- Craniocervical Instability and AAI
- GI Involvement in EDS
- POTS, MCAS and the EDS Triad
- Pain Management in hEDS
- Pediatric EDS and Transition of Care
- Physical Therapy and Joint Protection
- Pregnancy and EDS
- Hypertension
- Osteoporosis
- POTS
- POTS Subtypes
- MCAS
- Collagen