Pulmonary Hypertension
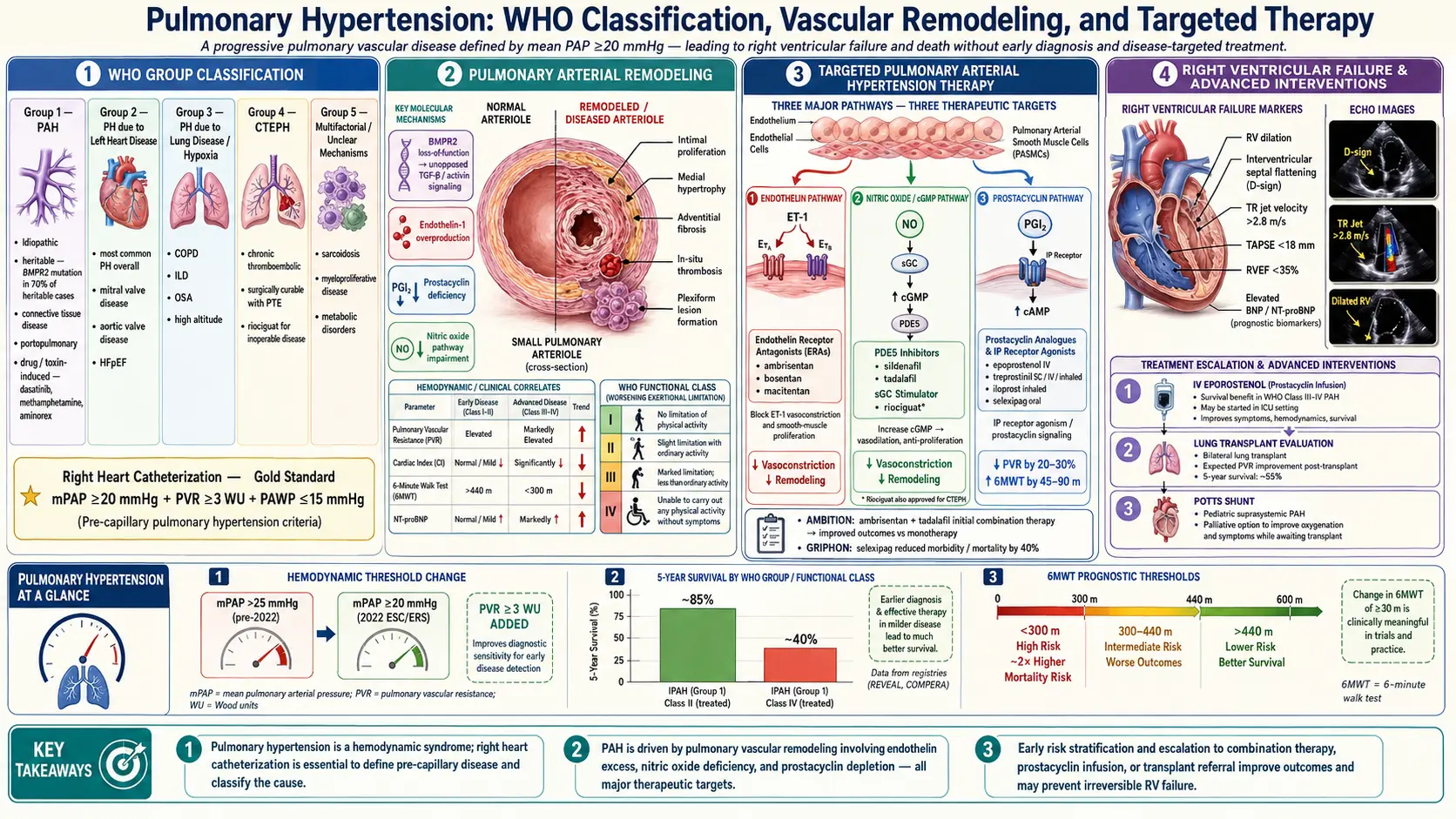
Pulmonary hypertension (PH) is a hemodynamic state defined by elevated mean pulmonary arterial pressure (mPAP) at rest. It encompasses a heterogeneous group of conditions unified by their effect on the pulmonary vasculature and the right ventricle, ultimately causing right heart failure if untreated. Modern classification into five WHO groups guides both diagnosis and therapy, with Group 1 pulmonary arterial hypertension (PAH) being the form with the most targeted pharmaceutical options.
Table of Contents
- Definition and WHO Classification
- Group 1: Pulmonary Arterial Hypertension (PAH)
- Groups 2–5: Other Forms of PH
- Pathophysiology and Right Heart Failure
- Symptoms and Clinical Presentation
- Diagnostic Workup
- Right Heart Catheterization: The Gold Standard
- Echocardiography
- PAH-Specific Therapy
- Upfront Combination Therapy (AMBITION Trial)
- CTEPH and Balloon Pulmonary Angioplasty
- Lung Transplantation
- References & Research
- Featured Videos
Definition and WHO Classification
Pulmonary hypertension is defined hemodynamically as a mean pulmonary arterial pressure (mPAP) greater than 20 mmHg measured by right heart catheterization (RHC) at rest. The 2022 ESC/ERS guidelines revised the threshold downward from the prior 25 mmHg, recognizing that any mPAP above 20 mmHg is associated with increased mortality.
The World Health Organization (WHO) classification divides PH into five groups based on pathophysiology, clinical presentation, and treatment approach. Accurate group assignment is essential because therapies that benefit one group may be harmful in another.
- Group 1 — Pulmonary Arterial Hypertension (PAH): Pre-capillary PH with primary pulmonary vascular disease; PCWP ≤15 mmHg; PVR ≥3 Wood units. Includes idiopathic, heritable, drug/toxin-induced, and associated forms (connective tissue disease, HIV, portal hypertension, congenital heart disease).
- Group 2 — PH due to Left Heart Disease: The most common form of PH overall. Post-capillary elevation from elevated left atrial pressure (PCWP >15 mmHg). Causes: HFpEF, HFrEF, valvular heart disease. PAH-specific drugs are not indicated and may be harmful.
- Group 3 — PH due to Lung Disease and/or Hypoxia: Chronic hypoxic vasoconstriction and vascular remodeling from COPD, ILD, OSA, high-altitude exposure. Treat the underlying lung disease and hypoxia first.
- Group 4 — Chronic Thromboembolic PH (CTEPH): Caused by organized thromboembolic obstruction of the pulmonary vascular bed after PE. Unique because it is potentially surgically curable by pulmonary endarterectomy (PEA). Lifelong anticoagulation is mandatory.
- Group 5 — PH with Unclear or Multifactorial Mechanisms: Hematologic disorders (chronic hemolytic anemias), systemic diseases (sarcoidosis), metabolic disorders (glycogen storage diseases), and fibrosing mediastinitis.
Group 1: Pulmonary Arterial Hypertension (PAH)
PAH is defined by the combination of pre-capillary PH (mPAP >20 mmHg, PCWP ≤15 mmHg) and elevated pulmonary vascular resistance (PVR ≥3 Wood units) in the absence of other causes of pre-capillary PH. The underlying lesion is progressive obliterative remodeling of the small pulmonary arterioles, including plexiform lesions, medial hypertrophy, intimal fibrosis, and in situ thrombosis.
Subtypes of Group 1 PAH
- Idiopathic PAH (IPAH): No identifiable cause. Predominantly affects young to middle-aged women (female-to-male ratio approximately 2–4:1).
- Heritable PAH (HPAH): Germline mutations, most commonly in BMPR2 (bone morphogenetic protein receptor type 2) — present in 70–80% of familial cases and 10–20% of sporadic IPAH. BMPR2 mutations reduce the anti-proliferative signaling that normally suppresses pulmonary arterial smooth muscle proliferation. Other mutations: ACVRL1, ENG, SMAD9, CAV1, KCNK3.
- Drug- and Toxin-Induced PAH: Definite association: aminorex (historical), fenfluramine/dexfenfluramine, dasatinib (tyrosine kinase inhibitor). Likely association: amphetamines, methamphetamine, cocaine.
- Associated PAH (APAH): Connective tissue diseases — especially systemic sclerosis (SSc), where PAH complicates up to 10–15% of cases and is a leading cause of death. HIV infection (~0.5% of HIV patients). Portopulmonary hypertension (PoPH) in 2–6% of patients with portal hypertension. Eisenmenger syndrome from congenital heart disease.
Groups 2–5: Other Forms of PH
Group 2: Left Heart Disease (Most Common Form of PH)
Left heart disease is by far the most prevalent cause of elevated pulmonary pressures in clinical practice. Heart failure with preserved ejection fraction (HFpEF) has become the dominant etiology. The key hemodynamic distinction: Group 2 PH has PCWP >15 mmHg (reflecting elevated left atrial pressure), whereas Group 1 PAH has PCWP ≤15 mmHg. This distinction can only be reliably made by RHC — echocardiographic estimates of PCWP are insufficiently accurate. PAH-specific drugs are not indicated for Group 2 and may cause harm by further unloading the left ventricle.
Group 3: Lung Disease and Hypoxia
Chronic hypoxia is a potent pulmonary vasoconstrictor. In COPD and ILD, hypoxic pulmonary vasoconstriction combined with destruction of the vascular bed elevates pulmonary pressures. Treatment is directed at the underlying lung disease and correction of hypoxia with supplemental oxygen. PAH-specific drugs have not shown benefit in unselected Group 3 PH.
Group 4: CTEPH
CTEPH develops in approximately 3–5% of patients following acute pulmonary embolism when clots fail to fully resolve and organize into fibrous obstructions. It is the only form of PH potentially curable by surgical intervention — pulmonary endarterectomy (PEA). All CTEPH patients require lifelong anticoagulation.
Pathophysiology and Right Heart Failure
Regardless of group, the final common pathway of PH is progressive elevation of pulmonary vascular resistance (PVR), forcing the right ventricle (RV) to generate increasing pressure to maintain cardiac output. The RV initially adapts through hypertrophy (concentric remodeling), but eventually dilates and fails. RV failure is the proximate cause of death in PAH.
Three Therapeutic Pathways in PAH
- Endothelin pathway: Endothelin-1 (ET-1) is overproduced in PAH, causing vasoconstriction and smooth muscle proliferation via ET-A receptors. Endothelin receptor antagonists (ERAs) block these receptors.
- Nitric oxide/cGMP pathway: Reduced NO bioavailability in PAH impairs vasodilation. PDE5 inhibitors prevent cGMP breakdown; sGC stimulators enhance cGMP production independent of NO.
- Prostacyclin pathway: Prostacyclin (PGI2) is a potent vasodilator and anti-proliferative mediator deficient in PAH. Prostacyclin analogues and IP receptor agonists restore this pathway.
Symptoms and Clinical Presentation
PH is often called the "silent killer" because symptoms are nonspecific and develop insidiously. The median time from symptom onset to diagnosis in IPAH historically has been 2–3 years.
- Exertional dyspnea: The most common and earliest symptom, reflecting reduced cardiac output on exertion.
- Fatigue: Related to reduced cardiac output and RV dysfunction.
- Syncope or near-syncope with exertion: A serious symptom indicating critically reduced cardiac output; associated with sudden death risk.
- Chest pain (angina-type): RV ischemia from RV pressure overload and reduced coronary perfusion.
- Palpitations: Atrial flutter and atrial fibrillation are common, exacerbated by RV dilation.
- Lower extremity edema: Sign of RV failure and elevated right atrial pressure; ascites may develop in severe cases.
- Hemoptysis: Occurs in a minority; potentially life-threatening in plexiform-lesion PAH or Eisenmenger syndrome.
Physical examination findings include: loud P2; right ventricular heave; tricuspid regurgitation murmur; elevated JVP; hepatomegaly; peripheral edema; and in Eisenmenger syndrome, central cyanosis and digital clubbing.
Diagnostic Workup
The diagnostic evaluation of PH has two goals: confirming elevated pulmonary pressures and determining the WHO group (etiology). A systematic stepwise approach is recommended by international guidelines.
Initial Evaluation
- Echocardiography: First-line screening test; estimates PASP from tricuspid regurgitation (TR) velocity. TR velocity >3.4 m/s has high probability of PH. Also evaluates RV size, function, and left heart disease.
- ECG: Right axis deviation, RBBB, P pulmonale, S1Q3T3 pattern.
- Chest X-ray: Enlarged central pulmonary arteries, pruning of peripheral vessels, RV enlargement.
- Pulmonary function tests: Screen for COPD or ILD as Group 3 causes. Isolated reduced DLCO suggests vascular disease.
- Overnight oximetry or polysomnography: Screen for sleep-disordered breathing as Group 3 contributor.
- V/Q scan: Mandatory in all newly diagnosed PH to screen for CTEPH — higher sensitivity than CT pulmonary angiography for peripheral segmental defects.
Connective Tissue and Other Workup
- ANA, anti-SCL-70, anti-centromere, anti-dsDNA — identify connective tissue disease-associated PAH
- HIV serology
- Liver function tests, hepatitis B/C serology, abdominal ultrasound — screen for portopulmonary hypertension
- BNP/NT-proBNP — marker of RV wall stress; elevated in PH, provides prognostic information
- Six-minute walk test (6MWT) — functional assessment; distance predicts outcomes
Right Heart Catheterization: The Gold Standard
Right heart catheterization (RHC) is mandatory before initiating PAH-specific therapy. Echocardiographic estimates of PASP are frequently inaccurate and cannot distinguish pre-capillary from post-capillary PH.
Key Hemodynamic Parameters
- mPAP >20 mmHg: Defines PH (2022 revised threshold)
- PCWP ≤15 mmHg: Pre-capillary PH (Groups 1, 3, 4, 5)
- PCWP >15 mmHg: Post-capillary PH (Group 2)
- PVR ≥3 Wood units (240 dyn·sec·cm⁻⁵): Required for Group 1 PAH diagnosis
- Cardiac index <2.5 L/min/m²: Sign of hemodynamic compromise
Acute Vasoreactivity Testing
In IPAH and HPAH, acute vasoreactivity testing with inhaled nitric oxide, IV adenosine, or IV epoprostenol identifies the minority (~10–15%) who are "vasoreactive" — mPAP decrease of ≥10 mmHg to ≤40 mmHg with maintained or increased cardiac output. Vasoreactive patients can be treated with high-dose calcium channel blockers (amlodipine, diltiazem, nifedipine) with sustained benefit in approximately 50% of this select group. Non-vasoreactive patients (the majority) do not benefit from CCBs.
Echocardiography
While not sufficient for diagnosis alone, echocardiography is indispensable throughout PH management — for initial detection, serial monitoring, and pre-/post-surgical assessment.
Key Echo Findings in PH
- TR jet velocity: Used with the simplified Bernoulli equation (PASP = 4v² + RAP) to estimate PASP.
- RV size and function: RV/LV basal diameter ratio >1.0 indicates RV dilation. TAPSE <17 mm indicates RV systolic dysfunction and portends worse prognosis.
- Interventricular septal flattening: D-shaped LV in systole/diastole indicates RV pressure/volume overload.
- Pericardial effusion: Present in up to 20% of PAH patients; associated with worse prognosis.
- IVC dilation without inspiratory collapse: Reflects elevated right atrial pressure (>10 mmHg).
PAH-Specific Therapy
Three pathways are targeted by approved PAH drugs. These drugs are indicated for Group 1 PAH only (and riociguat also for inoperable CTEPH). They are not indicated for — and may worsen — Groups 2 and 3 PH.
Endothelin Receptor Antagonists (ERAs)
- Bosentan (oral, twice daily) — dual ETA/ETB blocker; liver function monitoring required; teratogenic.
- Ambrisentan (oral, once daily) — selective ETA blocker; lower hepatotoxicity risk; peripheral edema is the most common side effect; teratogenic.
- Macitentan (oral, once daily) — dual ETA/ETB with tissue targeting; demonstrated mortality/morbidity benefit in the SERAPHIN trial; teratogenic.
PDE5 Inhibitors
- Sildenafil (oral, three times daily) — inhibits PDE5 to prevent cGMP breakdown; improves exercise capacity and hemodynamics in PAH (SUPER-1 trial). Contraindicated with organic nitrates.
- Tadalafil (oral, once daily) — longer-acting PDE5 inhibitor; once-daily dosing (PHIRST trial). Contraindicated with nitrates and riociguat.
Soluble Guanylate Cyclase (sGC) Stimulator
- Riociguat (oral, three times daily) — stimulates sGC and sensitizes it to endogenous NO, increasing cGMP independent of NO levels. Approved for both PAH (PATENT-1) and inoperable CTEPH (CHEST-1). Contraindicated with PDE5 inhibitors and nitrates.
Prostacyclin Pathway Agents
- Epoprostenol (IV): Continuous intravenous infusion via permanent central venous catheter; the only PAH drug with proven survival benefit in RCT; drug of choice for WHO FC IV. Life-threatening rebound PH if infusion is interrupted.
- Iloprost (inhaled): 6–9 times daily via nebulizer; short duration (~45–90 min per dose); useful for ambulatory patients with moderate disease.
- Treprostinil: Multiple formulations — subcutaneous (SC) continuous infusion, IV, inhaled (4 times daily), and oral (twice daily).
- Selexipag (oral): IP receptor agonist (not a prostacyclin analogue); once-daily selective IP1 receptor agonist; demonstrated reduction in morbidity/mortality in the GRIPHON trial — the largest PAH morbidity/mortality study conducted.
Upfront Combination Therapy (AMBITION Trial)
For newly diagnosed WHO FC II–III treatment-naive patients, upfront combination therapy targeting two or more pathways simultaneously is now guideline-recommended and represents the most important advance in PAH management in the past decade.
The AMBITION Trial (2015)
The AMBITION trial compared ambrisentan + tadalafil (initial combination) versus ambrisentan alone or tadalafil alone in treatment-naive patients with WHO FC II–III PAH. The combination arm achieved a 50% reduction in the primary composite endpoint of clinical failure events (death, hospitalization, disease progression, unsatisfactory long-term clinical response). This landmark trial shifted guidelines away from sequential add-on therapy toward initial combination treatment.
Escalation Strategy
For patients not at low risk at diagnosis, or those deteriorating despite dual therapy, sequential escalation to triple oral combination or addition of prostacyclin pathway agents (inhaled or SC) is appropriate. WHO FC IV patients should receive IV epoprostenol as the backbone of therapy given its survival benefit.
CTEPH and Balloon Pulmonary Angioplasty
Pulmonary Endarterectomy (PEA)
PEA is the treatment of choice for operable CTEPH. Performed under deep hypothermic circulatory arrest at specialized centers, PEA removes the organized fibrous obstructions from the pulmonary arterial tree. Five-year survival after successful PEA at experienced centers exceeds 80–90%. Surgical operability requires accessible proximal disease and acceptable PVR.
Balloon Pulmonary Angioplasty (BPA)
For inoperable CTEPH (distal disease or unacceptable surgical risk), balloon pulmonary angioplasty (BPA) is an important catheter-based intervention. Developed in Japan, BPA involves serial sessions (typically 4–8+) of balloon dilation of organized thromboembolic lesions in segmental and subsegmental pulmonary arteries. BPA significantly reduces PVR, mPAP, and improves 6MWT and RV function. The RACE trial (2020) compared BPA versus riociguat as initial therapy for inoperable CTEPH, demonstrating BPA superiority for hemodynamic outcomes. Riociguat can also be combined with BPA.
Lung Transplantation
Lung transplantation remains the ultimate option for patients with PAH refractory to maximal medical therapy. It is considered an endpoint rather than a primary treatment strategy. Patients should be referred to a transplant center early — typically when deteriorating on dual oral combination therapy before FC IV is established.
Bilateral lung transplantation is preferred over single lung for PAH, as single lung transplantation leaves an unbalanced vascular bed risking severe primary graft dysfunction. Heart-lung transplantation is reserved for complex congenital heart disease requiring concurrent cardiac repair. Five-year survival after bilateral lung transplant for PAH is approximately 50–55% at experienced centers.
References & Research
Key Research Papers
- Humbert M, Kovacs G, Hoeper MM, et al; ESC/ERS Scientific Document Group. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618-3731. PMID 36017548
- Galie N, Barbera JA, Frost AE, et al; AMBITION Investigators. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373(9):834-844. PMID 26308684
- McLaughlin VV, Sitbon O, Badesch DB, et al. Survival with first-line bosentan in patients with primary pulmonary arterial hypertension. Eur Respir J. 2005;25(2):244-249. PMID 15684287
- Ghofrani HA, Galie N, Grimminger F, et al; PATENT-1 Study Group. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330-340. — Search PubMed
- Ghofrani HA, D'Armini AM, Grimminger F, et al; CHEST-1 Study Group. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369(4):319-329. PMID 23883377
- Sitbon O, Channick R, Chin KM, et al; GRIPHON Investigators. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373(26):2522-2533. PMID 26699168
- Pulido T, Adzerikho I, Channick RN, et al; SERAPHIN Investigators. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809-818. PMID 23984728
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. PMID 30545968
- Kim NH, Delcroix M, Jais X, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J. 2019;53(1):1801915. PMID 30545971
- Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334(5):296-302. PMID 8532025
- Olschewski H, Simonneau G, Galie N, et al; Aerosolized Iloprost Randomized Study Group. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322-329. PMID 12151469
- Boucly A, Weatherald J, Savale L, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2017;50(2):1700248. — Search PubMed
Research Papers
The following PubMed topic searches retrieve current peer-reviewed literature on Pulmonary Hypertension. Each link opens a live PubMed query.
- Pulmonary arterial hypertension treatment
- PH right heart catheterization
- BMPR2 mutation pulmonary hypertension
- Endothelin receptor antagonist PAH
- Sildenafil pulmonary hypertension
- CTEPH pulmonary endarterectomy
- Balloon pulmonary angioplasty CTEPH
- PH left heart disease Group 2
- Scleroderma pulmonary hypertension
- Epoprostenol IV pulmonary arterial hypertension
Connections
- Pulmonology
- Heart Failure
- Pleural Effusion
- Pulmonary Embolism
- COPD
- Scleroderma
- Hypertension
- Interstitial Lung Disease
- Cardiology