Interstitial Lung Disease
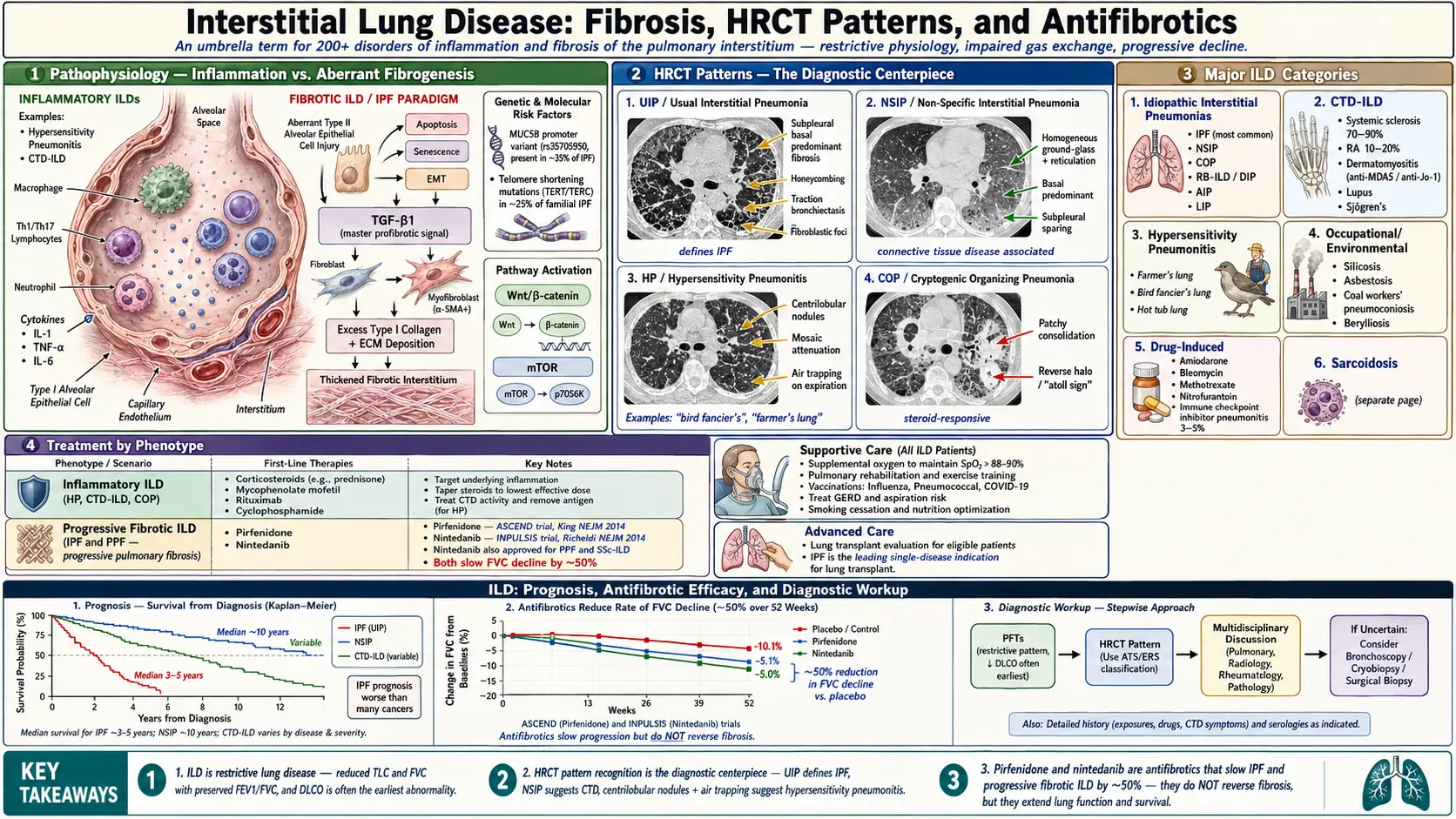
Table of Contents
- Overview
- Epidemiology
- Pathophysiology — Fibrosis Mechanisms
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis — PFTs, HRCT, Bronchoscopy, Surgical Biopsy
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research and Advances
- Research Papers
- Connections
- Featured Videos
1. Overview
Interstitial lung disease (ILD) is an umbrella term encompassing over 200 distinct disorders characterized by diffuse inflammation and/or fibrosis of the pulmonary interstitium — the tissue and space surrounding alveoli, capillaries, and bronchioles. Though structurally the interstitium is the primary target, ILDs invariably affect alveolar epithelium, capillary endothelium, and airspaces as well. The result is progressive impairment of gas exchange and lung mechanics, leading to restrictive physiology, hypoxemia, and functional decline.
The most clinically significant ILD is idiopathic pulmonary fibrosis (IPF), the prototype of the progressive fibrosing ILDs. Other major categories include connective tissue disease-associated ILD (CTD-ILD), hypersensitivity pneumonitis (HP), sarcoidosis, occupational/environmental ILDs, and drug-induced ILD. Accurate classification is critical because prognosis and treatment vary substantially among subtypes.
2. Epidemiology
The prevalence of all ILDs combined is estimated at 80–100 per 100,000 in the United States. IPF, the most common idiopathic IIP, has an estimated prevalence of 14–43 per 100,000 and incidence of 7–16 per 100,000 per year. IPF is predominantly a disease of older males (>60 years), with a strong association with cigarette smoking.
CTD-ILD is the most common ILD category overall; systemic sclerosis (SSc) is complicated by ILD in 70–90% of patients (though progressive disease in ~15%), and rheumatoid arthritis ILD affects approximately 10–20% of RA patients. Hypersensitivity pneumonitis accounts for up to 15% of ILD diagnoses in tertiary centers. Occupational ILDs (silicosis, asbestosis, coal worker's pneumoconiosis) remain significant public health concerns globally.
Drug-induced ILD is increasingly recognized with >350 implicated medications (bleomycin, amiodarone, methotrexate, nitrofurantoin, immune checkpoint inhibitors). ILD incidence is increasing, partly reflecting improved recognition and an aging population.
3. Pathophysiology — Fibrosis Mechanisms
The mechanisms underlying ILD vary by subtype, but converge on two principal processes: alveolar inflammation and/or aberrant fibrogenesis.
Inflammatory ILDs (e.g., Hypersensitivity Pneumonitis, CTD-ILD)
Repetitive exposure to antigenic or injurious stimuli triggers innate and adaptive immune responses. Alveolar macrophages, T lymphocytes (particularly Th1/Th17 in HP), and neutrophils accumulate in the interstitium. Macrophage-derived cytokines (IL-1, TNF-alpha, IL-6) amplify inflammation. In some cases, inflammation resolves; in others, it progresses to fibrosis via TGF-beta-mediated activation of fibroblasts and myofibroblasts.
Fibrotic ILDs — IPF Paradigm
IPF is driven by aberrant alveolar epithelial repair rather than primary inflammation. Repetitive micro-injuries (cigarette smoke, viral infections, gastroesophageal reflux microaspiration, mechanical stress in susceptible individuals) trigger abnormal type II alveolar epithelial cell (AEC II) responses: apoptosis, senescence, and epithelial-mesenchymal transition (EMT). Key molecular mechanisms:
- TGF-beta1 signaling: The master profibrotic mediator. Activates fibroblasts to differentiate into alpha-SMA-positive myofibroblasts, which produce excess extracellular matrix (ECM) — particularly type I collagen. Myofibroblasts are remarkably resistant to apoptosis.
- Wnt/beta-catenin pathway: Activated in IPF; promotes fibroblast proliferation and matrix production.
- mTOR pathway: Regulates fibroblast metabolism and autophagy; potential therapeutic target.
- Telomere biology: Telomere shortening mutations (TERT, TERC) found in ~25% of familial IPF and ~3% of sporadic IPF; impairs AEC II renewal capacity.
- Mucin gene variants: MUC5B promoter polymorphism (rs35705950) is the strongest genetic risk factor for IPF; present in ~35% of IPF patients vs ~10% of controls; mechanism involves abnormal mucus production impairing mucociliary defense.
Histologically, IPF demonstrates the usual interstitial pneumonia (UIP) pattern: temporally heterogeneous fibrosis (alternating areas of fibrosis and near-normal lung), fibroblastic foci (leading edges of active fibrosis), honeycomb changes (cystic airspace remodeling) predominantly in the subpleural, basal lung.
4. Etiology and Risk Factors
ILDs are classified by etiology:
Idiopathic Interstitial Pneumonias (IIPs)
- IPF (UIP pattern) — most common; progressive; no known cause
- Nonspecific interstitial pneumonia (NSIP) — often CTD-associated; better prognosis than IPF
- Cryptogenic organizing pneumonia (COP) — patchy consolidation; steroid-responsive
- Respiratory bronchiolitis-ILD (RB-ILD) / Desquamative interstitial pneumonia (DIP) — smoking-related
- Acute interstitial pneumonia (AIP/Hamman-Rich syndrome) — rapid progression; poor prognosis
- Lymphoid interstitial pneumonia (LIP) — often CTD or HIV-associated
Connective Tissue Disease-Associated ILD
- Systemic sclerosis (NSIP or UIP pattern)
- Rheumatoid arthritis (UIP most common)
- Polymyositis/dermatomyositis (NSIP; anti-synthetase syndrome: anti-Jo-1, anti-MDA5)
- Systemic lupus erythematosus, Sjogren's syndrome, MCTD
Hypersensitivity Pneumonitis
- Farmer's lung (thermophilic Actinomycetes)
- Bird fancier's lung (avian proteins)
- Hot tub lung (nontuberculous mycobacteria)
- Isocyanate-induced HP (occupational)
Occupational and Environmental ILDs
- Silicosis (crystalline silica) — accelerated silicosis risk with sandblasting, hydraulic fracturing
- Asbestosis (tremolite, chrysotile asbestos)
- Coal workers' pneumoconiosis
- Berylliosis (chronic beryllium disease)
- Hard metal lung disease (cobalt)
Drug-Induced ILD
- Amiodarone, bleomycin, methotrexate, nitrofurantoin, cyclophosphamide
- Immune checkpoint inhibitors (pembrolizumab, nivolumab): pneumonitis in 3–5% of treated patients
- Tyrosine kinase inhibitors (gefitinib, erlotinib)
Risk Factors for ILD in General
- Cigarette smoking (IPF, RB-ILD, DIP, combined pulmonary fibrosis and emphysema)
- Age >50 years (IPF predominantly a disease of the elderly)
- Male sex (IPF)
- Family history (familial IPF; telomerase gene mutations)
- Gastroesophageal reflux (microaspiration)
- Occupational/environmental antigen exposure
5. Clinical Presentation
ILD typically presents with an insidious onset of symptoms, often leading to significant delay in diagnosis (median 1–2 years for IPF).
Symptoms:
- Progressive exertional dyspnea (hallmark; present in virtually all patients)
- Dry, nonproductive cough (IPF); productive cough may suggest HP or infection
- Fatigue, weight loss (particularly CTD-ILD, malignancy-associated ILD)
- Constitutional symptoms (fever, myalgias) in inflammatory ILDs (HP, COP, acute ILD)
- Hemoptysis — uncommon; suggests DAH or malignancy
- Extrapulmonary features: joint symptoms (CTD), skin changes (scleroderma, DM), sicca symptoms (Sjogren's), proximal muscle weakness (myositis)
Physical examination findings:
- Fine, "velcro-like" inspiratory crackles at lung bases (IPF, asbestosis, UIP pattern)
- Absence of crackles in NSIP, HP (mid-field crackles possible)
- Digital clubbing (30–50% of IPF; rare in other IIPs)
- Cyanosis (late disease)
- Signs of pulmonary hypertension (loud P2, RV heave, peripheral edema) in advanced disease
- Skin, joint, or mucosal findings suggesting underlying CTD
- "Mechanic's hands" (anti-synthetase syndrome), Raynaud's phenomenon (SSc, MCTD)
6. Diagnosis — PFTs, HRCT, Bronchoscopy, Surgical Biopsy
Pulmonary Function Tests (PFTs)
The hallmark of ILD is a restrictive ventilatory defect:
- Reduced total lung capacity (TLC <80% predicted)
- Reduced FVC with preserved or elevated FEV1/FVC ratio
- Reduced diffusing capacity for carbon monoxide (DLCO) — often the earliest abnormality; reflects impaired alveolar-capillary membrane function
- Reduced lung compliance (increased work of breathing)
- Combined obstructive-restrictive pattern in combined pulmonary fibrosis and emphysema (CPFE): "normal" TLC despite fibrosis, with markedly reduced DLCO
FVC decline of ≥10% or DLCO decline ≥15% over 6–12 months predicts disease progression and mortality in IPF.
High-Resolution CT (HRCT)
The cornerstone of ILD diagnosis. CT patterns guide diagnosis and can obviate surgical biopsy:
- UIP pattern (IPF): Bilateral, peripheral, basal-predominant honeycombing with or without peripheral traction bronchiectasis; absence of features suggesting alternative diagnosis. Typical UIP on HRCT is diagnostic of IPF in the correct clinical context.
- NSIP pattern: Bilateral, symmetric, basal ground-glass opacity and reticulation; subpleural sparing (classic); traction bronchiectasis in fibrotic NSIP.
- Organizing pneumonia: Bilateral peripheral and/or peribronchial consolidation; migratory nature.
- Hypersensitivity pneumonitis: Upper/mid-lobe predominant ground-glass opacity; centrilobular nodules; air trapping on expiratory CT (mosaic attenuation); fibrosis in chronic HP mimicking UIP/NSIP.
- Sarcoidosis: Perilymphatic nodules along bronchovascular bundles; upper lobe predominance; lymphadenopathy.
- Desquamative IP/RB-ILD: Diffuse ground-glass opacity; upper lobe centrilobular nodules (RB-ILD).
Bronchoscopy with Bronchoalveolar Lavage (BAL)
BAL cellular analysis provides diagnostic clues:
- Lymphocytosis (>40%): HP, sarcoidosis, NSIP, COP
- Neutrophilia: IPF, acute exacerbation of ILD, bacterial infection
- Eosinophilia: Eosinophilic pneumonia, drug-induced ILD
- Hemosiderin-laden macrophages: Diffuse alveolar hemorrhage
- Transbronchial biopsy: useful for sarcoidosis, HP, COP; inadequate tissue for UIP/NSIP pattern characterization
- Cryobiopsy: larger samples than conventional forceps; intermediate between transbronchial and surgical biopsy; growing role in ILD diagnosis
Surgical Lung Biopsy (SLB)
Video-assisted thoracoscopic surgery (VATS) biopsy provides the largest tissue samples and remains the gold standard for histopathologic diagnosis when HRCT is non-diagnostic. Multiple lobes should be sampled. Operative mortality and morbidity risk must be weighed against diagnostic benefit; SLB may be deferred in elderly, physiologically compromised patients or where the clinical/CT presentation is sufficiently diagnostic.
Serological Testing
- ANA, anti-dsDNA, anti-Scl-70, anti-centromere, RF, anti-CCP — screen for CTD-ILD
- Anti-synthetase antibodies (Jo-1, PL-7, PL-12, EJ, OJ, MDA5) — inflammatory myopathy-ILD
- Specific IgG precipitins (avian, thermophilic bacteria) — HP serologies
- ACE level — sarcoidosis (low sensitivity/specificity; ~75%/90%)
- KL-6 (Krebs von den Lungen-6): Elevated in IPF and other fibrotic ILDs; used in Japan as a biomarker
- SP-D, SP-A: Surfactant proteins; prognostic biomarkers in IPF
Multidisciplinary Discussion (MDD)
Integration of clinical, radiological, and pathological data in a dedicated ILD multidisciplinary team (pulmonologist, thoracic radiologist, pathologist) is the recommended diagnostic approach and demonstrably improves diagnostic accuracy and inter-observer agreement.
7. Treatment
Treatment is highly ILD-subtype-specific. Inflammatory ILDs may respond to immunosuppression; fibrotic ILDs (particularly IPF) do not benefit and may worsen with corticosteroids.
Idiopathic Pulmonary Fibrosis
- Nintedanib (tyrosine kinase inhibitor targeting FGFR, PDGFR, VEGFR): INPULSIS trials showed reduction in FVC decline by ~50%; approved for IPF and progressive fibrosing ILDs
- Pirfenidone (anti-fibrotic, anti-inflammatory, anti-oxidant): ASCEND and CAPACITY trials showed reduction in FVC decline; approved for IPF
- Antifibrotics do not improve lung function; they slow progression
- Avoid immunosuppression (prednisone + azathioprine + N-acetylcysteine shown to be harmful in PANTHER-IPF)
- Manage comorbidities: GERD treatment (omeprazole), PH treatment, OSA treatment
- Pulmonary rehabilitation
- Supplemental oxygen for resting or exertional hypoxemia
- Lung transplantation for eligible patients (bilateral preferred; median post-transplant survival ~5 years in IPF)
- Palliative care and advance care planning
CTD-ILD
- SSc-ILD: Nintedanib (SENSCIS trial: 44% reduction in FVC decline); mycophenolate mofetil; cyclophosphamide; tocilizumab for rapidly progressive SSc-ILD
- RA-ILD: Mycophenolate mofetil, azathioprine; avoid methotrexate (ILD risk); rituximab for refractory cases
- Inflammatory myopathy-ILD: High-dose corticosteroids + calcineurin inhibitor (tacrolimus) for anti-synthetase syndrome; rituximab for refractory cases
Hypersensitivity Pneumonitis
- Antigen avoidance — the most important intervention; can result in stabilization or improvement in early HP
- Systemic corticosteroids (prednisone 0.5–1 mg/kg/day) for acute/subacute HP with significant respiratory compromise; tapered over 4–6 months
- Mycophenolate mofetil, azathioprine for steroid-sparing in chronic HP
- Nintedanib for progressive fibrosing chronic HP (INBUILD trial data support use in progressive fibrosis regardless of underlying ILD subtype)
Organizing Pneumonia
- Prednisone 0.75–1 mg/kg/day for 4–6 weeks, then taper over 6–12 months; rapid clinical and radiological response typical
- Relapse common (30–50%) upon steroid tapering; re-treat with corticosteroids
8. Complications
- Acute exacerbation of IPF (AE-IPF): Sudden, unexplained acceleration of respiratory failure superimposed on stable IPF; bilateral new ground-glass opacities; in-hospital mortality 50–80%; may be triggered by infection, aspiration, procedures, or be idiopathic
- Pulmonary hypertension: Develops in 30–85% of IPF patients; worsens prognosis; inhaled treprostinil (INCREASE trial) recently approved for PH-ILD
- Lung cancer: Risk elevated in IPF (particularly squamous cell and adenocarcinoma); cigarette smoking synergism; annual LDCT screening recommended
- Venous thromboembolism: Elevated risk; prophylaxis in hospitalized ILD patients
- Infections: Opportunistic infections with immunosuppressive therapy; pneumocystis jirovecii prophylaxis with significant immunosuppression
- Pneumothorax: Spontaneous pneumothorax from ruptured honeycomb cysts; can precipitate acute respiratory failure
- GERD: Common comorbidity in IPF; microaspiration may perpetuate fibrosis; antacid therapy recommended
- Depression and anxiety: Common; impair quality of life and exercise capacity; warrant specific attention and palliative support
9. Prognosis
Prognosis is highly subtype-dependent:
- IPF: Median survival 2–5 years from diagnosis without antifibrotic therapy; antifibrotics extend survival but do not cure the disease. 5-year survival approximately 20–40%. FVC <70% predicted and DLCO <40% predicted are associated with significantly worse prognosis.
- NSIP (idiopathic): Better prognosis than IPF; 5-year survival ~70–80% for cellular NSIP; fibrotic NSIP ~45–90% depending on extent of fibrosis.
- CTD-ILD: Variable; SSc-ILD 10-year survival ~55%; RA-ILD median survival ~2–3 years from ILD diagnosis in studies (better in recent cohorts with modern therapy).
- COP: Excellent prognosis with corticosteroid treatment; 5-year survival >80%.
- Chronic HP: Variable; fibrotic HP with UIP pattern behaves similarly to IPF; non-fibrotic HP with antigen avoidance may improve or stabilize.
The GAP (Gender, Age, Physiology) Index and Gender-Age-Physiology (GAP) staging system (incorporating FVC and DLCO) has been validated to predict 1-, 2-, and 3-year mortality in IPF and other fibrotic ILDs.
10. Prevention
- Smoking cessation: Reduces risk of IPF, RB-ILD, DIP, and combined pulmonary fibrosis and emphysema
- Occupational exposure reduction: Engineering controls, personal protective equipment (N95 respirators), regulatory exposure limits for silica, asbestos, beryllium, and other fibrogenic dusts
- Medical surveillance programs for workers with recognized occupational ILD risks
- Antigen avoidance in HP-susceptible individuals (bird fanciers, farmers)
- Drug-induced ILD prevention: Baseline pulmonary evaluation before initiating fibrogenic drugs; monitoring protocols for bleomycin (cumulative dose), amiodarone, methotrexate
- GERD management: Aggressive acid suppression to minimize microaspiration in IPF patients
- Genetic counseling for familial IPF with identified causative mutations
11. Recent Research and Advances
INBUILD Trial: Demonstrated that nintedanib reduces FVC decline in progressive fibrosing ILDs other than IPF (including NSIP, HP, RA-ILD, SSc-ILD), establishing the concept of a common progressive fibrosis phenotype amenable to antifibrotic therapy regardless of specific ILD diagnosis.
Inhaled Treprostinil (INCREASE Trial): Approved in 2021 for pulmonary hypertension associated with ILD; a significant advance as the first approved therapy specifically for PH-ILD, demonstrating improvement in 6MWD and reduction in clinical worsening.
Cryobiopsy: Transbronchial cryobiopsy is gaining acceptance as a less invasive alternative to surgical lung biopsy. Meta-analyses show comparable diagnostic yield with lower morbidity, though pneumothorax and bleeding rates require attention.
Genomic and Transcriptomic Profiling: Gene expression profiling (e.g., Envisia genomic classifier, Percepta registry) of BAL and transbronchial biopsy samples can distinguish UIP from non-UIP patterns non-invasively, potentially reducing need for surgical biopsy.
Senescence and Epigenetic Targeting: Senolytic agents (navitoclax, dasatinib + quercetin) targeting senescent AEC II cells are in early-phase clinical trials for IPF. Epigenetic modifications (histone deacetylase inhibition) represent another emerging therapeutic strategy.
Artificial Intelligence in HRCT: Deep learning algorithms can identify UIP and NSIP patterns on HRCT with accuracy approaching expert thoracic radiologists, and can quantify fibrosis extent to track progression.
GLP-1 Receptor Agonists in ILD: Preliminary data suggests metabolic comorbidities (obesity, diabetes) accelerate ILD progression; GLP-1 agonists may have anti-fibrotic properties beyond glycemic control.
12. References
- Raghu G, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205(9):e18–e47.
- Travis WD, et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–748.
- Richeldi L, et al. (INPULSIS). Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N Engl J Med. 2014;370(22):2071–2082.
- King TE, et al. (ASCEND). A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N Engl J Med. 2014;370(22):2083–2092.
- Flaherty KR, et al. (INBUILD). Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med. 2019;381(18):1718–1727.
- Distler O, et al. (SENSCIS). Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. N Engl J Med. 2019;380(26):2518–2528.
- Raghu G, et al. Diagnosis of Idiopathic Pulmonary Fibrosis: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68.
- Ley B, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684–691.
- Cottin V, et al. Syndrome of combined pulmonary fibrosis and emphysema. Eur Respir J. 2005;26(4):586–593.
- Idiopathic Pulmonary Fibrosis Clinical Research Network. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis (PANTHER-IPF). N Engl J Med. 2012;366(21):1968–1977.
- Waxman A, et al. (INCREASE). Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N Engl J Med. 2021;384(4):325–334.
- Ryu JH, et al. Hypersensitivity pneumonitis: a comprehensive review. J Investig Allergol Clin Immunol. 2020;30(2):78–95.
- Seibold MA, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503–1512.
- Collard HR, et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis: An International Working Group Report. Am J Respir Crit Care Med. 2016;194(3):265–275.
- Johannson KA, et al. Acute exacerbation of idiopathic pulmonary fibrosis: a review of current and novel pharmacotherapies. Eur Respir J. 2013;41(6):1464–1475.
- Cottin V, et al. Interstitial lung diseases associated with connective tissue diseases. Eur Respir Rev. 2016;25(140):213–228.
Research Papers
The following PubMed topic searches surface the current peer-reviewed literature on Interstitial Lung Disease. Each link opens a live PubMed query; results update as new papers are indexed.
- PubMed search: interstitial lung disease
- PubMed search: idiopathic pulmonary fibrosis
- PubMed search: pirfenidone pulmonary fibrosis
- PubMed search: nintedanib pulmonary fibrosis
- PubMed search: hypersensitivity pneumonitis
- PubMed search: nonspecific interstitial pneumonia
- PubMed search: cryptogenic organizing pneumonia
- PubMed search: connective tissue disease interstitial lung
- PubMed search: ILD high resolution CT
- PubMed search: lung transplantation pulmonary fibrosis
- PubMed search: progressive pulmonary fibrosis
- PubMed search: ILD bronchoalveolar lavage
Connections
- Pulmonology
- Sarcoidosis
- Pulmonary Hypertension
- Rheumatoid Arthritis
- Acute Respiratory Distress Syndrome
- Vitamin D3
- Shortness of Breath
- Chronic Cough
- Pneumonia
- Pulmonary Embolism
- Lupus
- Sjogren's Syndrome
- Fatigue
- Heart Failure
- Quercetin
- Cancer
- Diabetes
- COPD
- Inflammatory Markers
- Lung Cancer
- Rheumatoid Arthritis
- Pulmonary Fibrosis — the scarring end-stage pattern shared by the progressive fibrosing ILDs, including IPF.