Glomerulonephritis
Glomerulonephritis (GN) is a heterogeneous group of inflammatory disorders affecting the glomeruli of the kidney, resulting in hematuria, proteinuria, hypertension, and varying degrees of renal impairment. It is a leading cause of end-stage kidney disease (ESKD) worldwide and encompasses both primary (idiopathic) and secondary (systemic disease-associated) forms.
Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research and Advances
- References
1. Overview
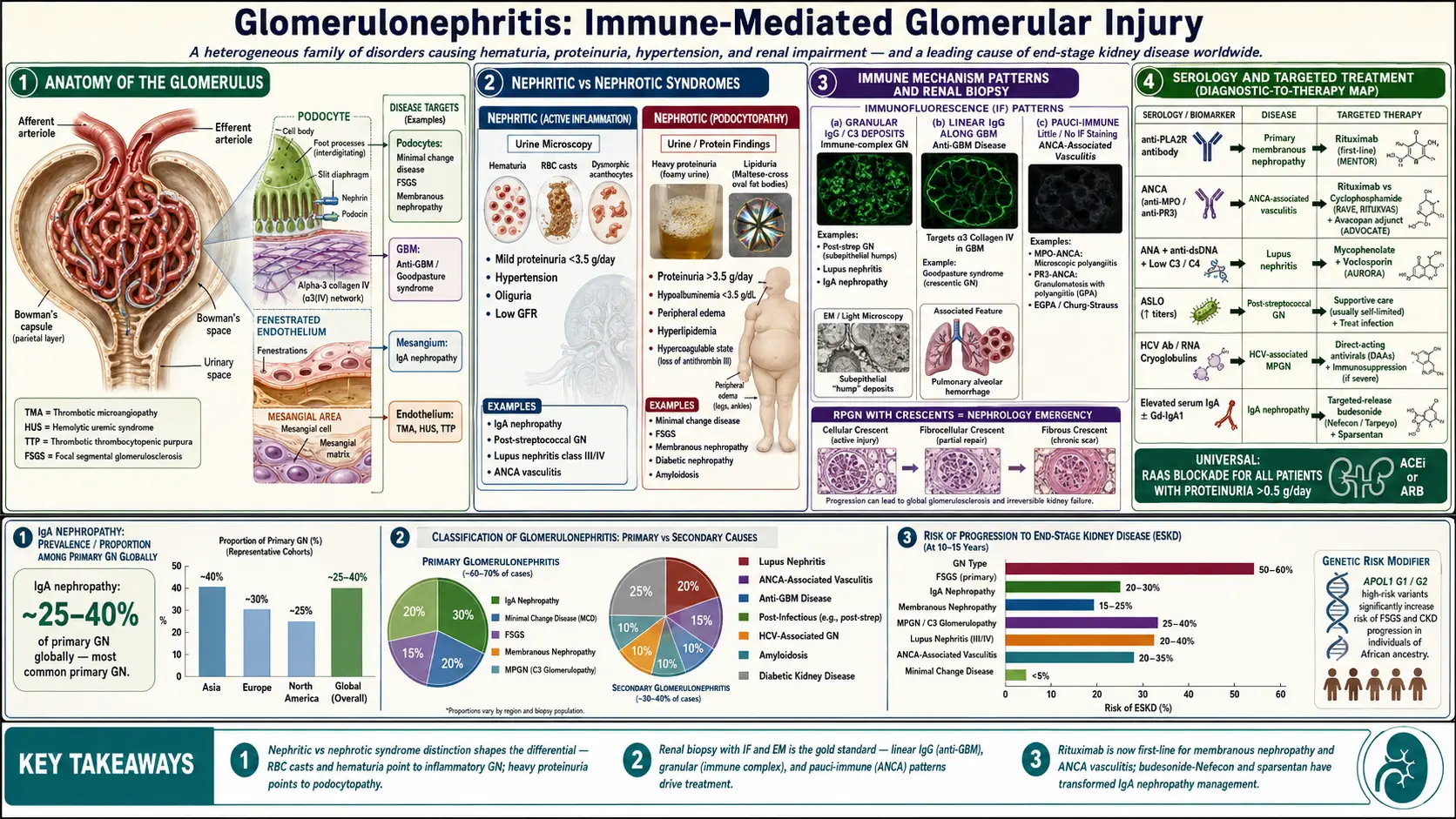
Glomerulonephritis encompasses a broad spectrum of glomerular diseases characterized by immune-mediated glomerular injury. It is clinically classified into distinct syndromes: the nephritic syndrome (hematuria, RBC casts, hypertension, oliguria, mild-to-moderate proteinuria) and the nephrotic syndrome (heavy proteinuria >3.5 g/day, hypoalbuminemia, edema, hyperlipidemia), though overlap is common.
A particularly severe form, rapidly progressive glomerulonephritis (RPGN), is defined by a rapid deterioration of kidney function (loss of ≥50% GFR within weeks to months) with crescentic changes on biopsy. RPGN is a nephrology emergency requiring urgent diagnosis and immunosuppressive therapy. GN is classified pathologically (by light microscopy, immunofluorescence, and electron microscopy) and etiologically.
2. Epidemiology
GN accounts for approximately 10–15% of all ESKD in developed countries and up to 30% globally. IgA nephropathy is the most common primary GN worldwide, with a prevalence of 25–40% of primary GN biopsies in Europe, Asia, and North America. Membranous nephropathy is the most common cause of nephrotic syndrome in adults over 40 in Western countries.
Post-infectious GN (PIGN) predominantly affects children aged 5–12 years in developing nations, most commonly following group A streptococcal pharyngitis or impetigo. ANCA-associated vasculitis (AAV) has an annual incidence of 20 per million population, predominantly affecting adults over 50. Lupus nephritis (LN) affects up to 60% of patients with systemic lupus erythematosus (SLE) and disproportionately impacts women of childbearing age and minority populations.
3. Pathophysiology
GN results from immune-mediated glomerular injury through several distinct mechanisms:
Immune Complex-Mediated GN
Circulating antigen-antibody complexes deposit in the glomerular mesangium, subendothelial, or subepithelial space, or antibodies react in situ with planted antigens. Complement activation (predominantly via the classical pathway) generates C3a and C5a (anaphylatoxins), the membrane attack complex (MAC, C5b-9), and recruits neutrophils and macrophages. This results in oxidative stress, protease release, and glomerular basement membrane (GBM) injury. Examples: post-infectious GN (subepithelial "humps"), lupus nephritis (subendothelial deposits in class III/IV), IgA nephropathy (mesangial deposits).
Anti-GBM Disease (Goodpasture Syndrome)
Autoantibodies directed against the alpha-3 chain of type IV collagen (alpha-3[IV]NC1) in the GBM and alveolar basement membrane cause linear IgG deposition on immunofluorescence. This results in rapidly progressive crescentic GN and, when pulmonary involvement occurs, diffuse alveolar hemorrhage. Genetic susceptibility involves HLA-DR15.
Pauci-Immune GN (ANCA-Associated Vasculitis)
MPO-ANCA or PR3-ANCA activate primed neutrophils, causing degranulation and endothelial injury without significant immunoglobulin deposition (pauci-immune on immunofluorescence). Microscopic polyangiitis (MPA), granulomatosis with polyangiitis (GPA, formerly Wegener's), and eosinophilic GPA (EGPA, formerly Churg-Strauss) are the three AAV subtypes. Crescentic GN with fibrinoid necrosis is the characteristic renal lesion.
Podocytopathy
In minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS), podocyte injury with effacement of foot processes is the primary pathological event, predominantly manifesting as nephrotic syndrome with variable GFR impairment. T-cell dysfunction and circulating permeability factors (suPAR, cardiotrophin-like cytokine-1) have been implicated in primary FSGS.
Complement-Mediated GN
C3 glomerulopathy (C3GN and dense deposit disease/MPGN type II) results from dysregulation of the alternative complement pathway (mutations in CFH, CFI, C3, CFB, or antibodies against factor H or C3 convertase). Membranoproliferative GN (MPGN) type I is immune complex-mediated; types II and III are now reclassified under C3 glomerulopathy.
4. Etiology and Risk Factors
Primary GN
- IgA nephropathy (Berger's disease): Galactose-deficient IgA1 (Gd-IgA1) overproduction and autoantibody-mediated mesangial deposition
- Minimal change disease (MCD): Idiopathic (most common in children); secondary to NSAIDs, lymphoma (especially Hodgkin's)
- Focal segmental glomerulosclerosis (FSGS): Primary (circulating permeability factors), secondary (hyperfiltration, HIV, heroin, obesity, reflux nephropathy), genetic (podocin, nephrin, TRPC6 mutations)
- Membranous nephropathy (MN): Primary (anti-PLA2R antibodies in 70–80%; anti-THSD7A in 3–5%); secondary (malignancy, hepatitis B, drugs, SLE)
- MPGN: Immune complex-mediated, C3 glomerulopathy, monoclonal immunoglobulin deposition disease
Secondary GN
- Lupus nephritis: SLE, classified by ISN/RPS 2018 into classes I–VI
- ANCA-associated vasculitis: GPA, MPA, EGPA
- Anti-GBM disease: Idiopathic; triggered by hydrocarbon inhalation, smoking, infections in genetically susceptible individuals
- Post-infectious GN: Streptococcal (pharyngitis, impetigo), staphylococcal (MRSA-associated GN), viral (hepatitis B/C, HIV)
- Cryoglobulinemic GN: Hepatitis C-associated mixed cryoglobulinemia (type II); monoclonal gammopathy
- Henoch-Schonlein Purpura (IgA vasculitis): Systemic IgA-mediated vasculitis
- Diabetic nephropathy and Amyloidosis (AL, AA)
Risk Factors
- Genetic predisposition: HLA alleles (HLA-DQ alleles in MN, HLA-DR15 in anti-GBM), APOL1 variants (G1/G2) significantly increase FSGS and ESKD risk in individuals of African ancestry
- Infections, autoimmune diseases, malignancies, medications
- Obesity, hypertension, smoking, illicit drug use
5. Clinical Presentation
Nephritic Syndrome
- Gross or microscopic hematuria (smoky or tea-colored urine), RBC casts on microscopy
- Hypertension (sodium and water retention, RAAS activation)
- Oliguria, edema (periorbital, peripheral)
- Mild-to-moderate proteinuria (typically <3.5 g/day)
- Elevated serum creatinine, reduced GFR
Nephrotic Syndrome
- Heavy proteinuria (>3.5 g/day), frothy urine
- Hypoalbuminemia (<3.5 g/dL), peripheral edema, ascites, pleural effusions
- Hyperlipidemia (elevated LDL, total cholesterol, triglycerides)
- Lipiduria (oval fat bodies, Maltese cross appearance under polarized light)
- Hypercoagulable state (loss of antithrombin III, protein C and S; venous thromboembolism and renal vein thrombosis in membranous nephropathy)
RPGN Features
- Rapidly worsening renal function (weeks to months), often with hematuria and oliguria
- Constitutional symptoms: fever, malaise, weight loss (particularly with vasculitis)
- Pulmonary-renal syndrome: hemoptysis + GN (anti-GBM disease, ANCA vasculitis, SLE)
- Upper respiratory tract symptoms (saddle-nose deformity, sinusitis in GPA)
6. Diagnosis
Urinalysis and Urine Microscopy
- Dipstick and microscopy: hematuria (dysmorphic red cells, acanthocytes), RBC casts (pathognomonic for GN), granular casts, heavy proteinuria
- 24-hour urine protein or spot urine protein-to-creatinine ratio (PCR): quantify proteinuria
Serum Biomarkers
- Creatinine and eGFR: Baseline and serial monitoring
- Albumin, lipid panel: Assess nephrotic syndrome
- ANCA serology: MPO-ANCA (perinuclear pattern, p-ANCA) and PR3-ANCA (cytoplasmic pattern, c-ANCA); ELISA preferred over immunofluorescence
- Anti-GBM antibodies: ELISA against alpha-3(IV)NC1
- Anti-PLA2R antibodies: Highly specific for primary membranous nephropathy (sensitivity 70–80%, specificity >99%); titer correlates with disease activity
- ANA, anti-dsDNA, complement (C3, C4, CH50): Lupus nephritis (low C3/C4 with active disease)
- ASLO, anti-DNase B: Post-streptococcal GN
- Hepatitis B surface antigen/antibody, hepatitis C RNA, HIV: Viral-associated GN
- Cryoglobulins, serum protein electrophoresis, free light chains: Cryoglobulinemic GN, monoclonal gammopathy
- C3, C4, CH50, factor H, factor I levels: C3 glomerulopathy workup
Kidney Biopsy
Renal biopsy is the gold standard for diagnosis and is essential in most adult patients with GN. It guides immunosuppressive therapy and provides prognostic information. Evaluation requires:
- Light microscopy: Identifies cellular pattern (diffuse, focal, segmental, global), mesangial expansion, crescents (cellular, fibrocellular, fibrous), necrosis, and sclerosis
- Immunofluorescence (IF): IgG, IgA, IgM, C3, C1q, kappa, lambda distribution and pattern (mesangial, subepithelial, subendothelial); linear IgG = anti-GBM; granular = immune complex; negative/pauci = ANCA
- Electron microscopy (EM): Location and character of electron-dense deposits; podocyte foot process effacement; GBM thickness; lamina densa alterations in dense deposit disease
Imaging
- Renal ultrasound: assess kidney size, echogenicity, symmetry; exclude obstruction
- Chest imaging: pulmonary infiltrates in pulmonary-renal syndromes
7. Treatment
General Measures
- Blood pressure control: target <130/80 mmHg; ACE inhibitors or ARBs preferred for antiproteinuric effect
- Dietary sodium restriction (<2 g/day) and fluid management for edema
- Statin therapy for hyperlipidemia in nephrotic syndrome
- Anticoagulation with warfarin or heparin for serum albumin <2.5 g/dL in membranous nephropathy with high thrombotic risk (MN-PRISM score)
- Diuretics for edema management; caution to avoid intravascular volume depletion
IgA Nephropathy
- RAAS blockade for all patients with proteinuria >0.5 g/day
- Targeted-release budesonide (Nefecon/Tarpeyo): approved by FDA for IgA nephropathy with urine PCR >1.5 g/g (NefIgArd trial); targets Peyer's patches to reduce Gd-IgA1
- Systemic corticosteroids for eGFR >30 mL/min/1.73 m² with persistent proteinuria >1 g/day (STOP-IgAN, TESTING trials guide individualized approach)
- Sparsentan (dual endothelin-angiotensin receptor antagonist): FDA-approved accelerated approval based on PROTECT trial proteinuria reduction
Minimal Change Disease
- Prednisone 1 mg/kg/day (max 80 mg) for 4–16 weeks; 80–90% achieve complete remission
- Cyclosporine or tacrolimus for frequently relapsing/steroid-dependent disease
- Rituximab for steroid-dependent or frequently relapsing MCD (MENTOR trial data)
Membranous Nephropathy
- Conservative management (RAAS blockade, statins, anticoagulation) for low-risk patients
- Rituximab is now first-line immunosuppression based on MENTOR trial (rituximab vs. cyclosporine); superior 24-month complete remission rates
- Cyclophosphamide + corticosteroids (Ponticelli regimen) for rituximab-refractory or unavailable settings
- Monitor anti-PLA2R titers to guide treatment response
ANCA-Associated Vasculitis
- Induction: Rituximab (375 mg/m² weekly × 4 or 1000 mg × 2) + high-dose corticosteroids; non-inferior to cyclophosphamide (RAVE and RITUXVAS trials) and superior for relapsing disease
- Cyclophosphamide IV pulse or oral for severe/life-threatening disease or rituximab unavailability
- Plasma exchange: not recommended routinely (PEXIVAS trial showed no mortality benefit); may be considered for dialysis-dependent patients
- Avacopan (C5a receptor inhibitor): FDA-approved as adjunct to reduce corticosteroid exposure (ADVOCATE trial)
- Maintenance: Rituximab 500 mg every 6 months for 18–24 months (MAINRITSAN trial); azathioprine or mycophenolate mofetil as alternatives
Anti-GBM Disease
- Plasma exchange (4 L daily for 14 days or until antibody undetectable) to remove circulating anti-GBM antibodies
- High-dose corticosteroids (methylprednisolone IV 500–1000 mg × 3 days, then prednisone 1 mg/kg/day)
- Cyclophosphamide 2–3 mg/kg/day for 3 months to prevent antibody rebound
- Dialysis support as required; kidney transplantation deferred until anti-GBM antibodies undetectable for ≥6 months
Lupus Nephritis
- Class III/IV (proliferative LN): mycophenolate mofetil (MMF) 3 g/day + low-dose corticosteroids (Euro-Lupus protocol) preferred over IV cyclophosphamide for most patients
- Voclosporin (calcineurin inhibitor) + MMF + low-dose steroids: AURORA 1 trial demonstrated superior complete renal response; FDA-approved for active LN
- Belimumab (anti-BLyS/BAFF): FDA-approved for active LN class III/IV/V (BLISS-LN trial)
- Class V (membranous LN): MMF + corticosteroids; consider tacrolimus or rituximab for refractory disease
- Hydroxychloroquine for all LN patients without contraindication
8. Complications
- Progression to CKD and ESKD: Risk varies by GN type and treatment response; FSGS and MPGN carry highest risk
- Hypertension: Major risk factor for accelerated GFR loss and cardiovascular events
- Thromboembolic disease: Renal vein thrombosis in membranous nephropathy (10–40%); DVT, pulmonary embolism
- Infections: Immunosuppression-related; encapsulated organisms (pneumococcal prophylaxis recommended); PCP prophylaxis with cyclophosphamide/rituximab (trimethoprim-sulfamethoxazole)
- Cardiovascular disease: Leading cause of mortality in ESKD; hyperlipidemia, hypertension, uremia-related accelerated atherosclerosis
- Drug toxicity: Cyclophosphamide (hemorrhagic cystitis, gonadal toxicity, malignancy); corticosteroids (osteoporosis, diabetes, infection); calcineurin inhibitors (nephrotoxicity, hypertension)
- AKI: RPGN, crescentic GN requiring emergent RRT
- Featured Videos
9. Prognosis
Prognosis is highly variable by GN subtype, treatment response, and degree of chronicity on biopsy. Key prognostic indicators include degree of proteinuria reduction, rate of GFR decline, hypertension control, and the proportion of globally sclerosed glomeruli and interstitial fibrosis on biopsy.
- IgA nephropathy: 20–30% progress to ESKD within 20 years; the IIgA-NS (Oxford MEST-C score) and International IgAN Prediction Tool guide risk stratification
- Membranous nephropathy: "Rule of thirds" — one-third spontaneous complete remission, one-third partial remission, one-third progress to ESKD; anti-PLA2R titer predicts spontaneous remission
- ANCA vasculitis: 5-year patient survival ~80%; renal relapse in 40–50% within 5 years; eGFR at presentation is the strongest predictor of renal survival
- Anti-GBM disease: Renal recovery unlikely if creatinine >6 mg/dL or anuria at presentation; overall patient survival improved with immunosuppression but dialysis dependence common
- Lupus nephritis: 10-year renal survival 60–85% depending on class; Black and Hispanic patients with LN have worse renal outcomes
- MCD: Excellent prognosis in children (95% remission); adults have higher relapse rates but rarely progress to ESKD
10. Prevention
- Early and adequate treatment of streptococcal infections to prevent post-streptococcal GN
- Antiviral therapy (entecavir, tenofovir) for HBV-associated GN
- Hepatitis C treatment with direct-acting antivirals (DAAs) resolves cryoglobulinemic GN in most cases
- Avoidance of nephrotoxic drugs and contrast media in patients with pre-existing GN
- Aggressive RAAS blockade to slow GFR decline and reduce proteinuria
- Hydroxychloroquine for lupus patients to reduce LN flares
- Regular monitoring and relapse surveillance in patients with treated GN
11. Recent Research and Advances
- Sparsentan: First-in-class dual endothelin-angiotensin receptor antagonist; FDA accelerated approval for IgA nephropathy (PROTECT trial, 2023); full approval pending eGFR endpoint confirmation
- Targeted-release budesonide (Nefecon): FDA-approved for IgA nephropathy (NefIgArd 2-year data showing sustained eGFR protection)
- Complement inhibition: Iptacopan (factor B inhibitor) achieved complete proteinuria response in primary membranous nephropathy and C3 glomerulopathy in phase II trials; pegcetacoplan for C3G under investigation
- Avacopan: C5aR1 inhibitor approved for ANCA vasculitis (ADVOCATE trial, 2021); replaces high-dose corticosteroids in induction, reducing glucocorticoid-associated toxicity
- CAR-T cell therapy: Early case reports and trials of anti-CD19 CAR-T cells for refractory SLE and ANCA vasculitis showing deep immunological remission
- SGLT2 inhibitors in GN: Dapagliflozin and empagliflozin reduce proteinuria and slow eGFR decline in IgA nephropathy and FSGS independent of diabetes (DAPA-CKD, EMPA-KIDNEY subgroup analyses)
- Anti-PLA2R monitoring: Serial antibody titers now guide treatment decisions and predict relapse in membranous nephropathy, enabling precision medicine approaches
12. References
- KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney International. 2021;100(4S):S1–S276.
- Rovin BH, Adler SG, Barratt J, et al. Executive summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney International. 2021;100(4):753–779.
- Bomback AS, Appel GB. Updates on the treatment of lupus nephritis. Journal of the American Society of Nephrology. 2010;21(12):2028–2035.
- Fervenza FC, Appel GB, Barbour SJ, et al. Rituximab or cyclosporine in the treatment of membranous nephropathy (MENTOR). New England Journal of Medicine. 2019;381(1):36–46.
- Rovin BH, Teng YKO, Ginzler EM, et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1). Lancet. 2021;397(10289):2070–2080.
- Furie RA, Rovin BH, Houssiau F, et al. Two-year, randomized, controlled trial of belimumab in lupus nephritis (BLISS-LN). New England Journal of Medicine. 2020;383(12):1117–1128.
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis (RAVE). New England Journal of Medicine. 2010;363(3):221–232.
- Jayne DRW, Merkel PA, Schall TJ, Bekker P. Avacopan for the treatment of ANCA-associated vasculitis (ADVOCATE). New England Journal of Medicine. 2021;384(7):599–609.
- Barratt J, Rovin BH, Cattran D, et al. Sparsentan in patients with IgA nephropathy: primary outcomes of the PROTECT randomized controlled trial. Lancet. 2023;401(10388):1584–1594.
- Lv J, Wong MG, Hladunewich MA, et al. Effect of oral methylprednisolone on clinical outcomes in patients with IgA nephropathy (TESTING). JAMA. 2022;327(19):1888–1898.
- Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis — a new look at an old entity. New England Journal of Medicine. 2012;366(12):1119–1131.
- Ponticelli C, Glassock RJ. Glomerular diseases: membranous nephropathy — a modern view. Clinical Journal of the American Society of Nephrology. 2014;9(3):609–616.
- Walsh M, Merkel PA, Peh CA, et al. Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis (PEXIVAS). New England Journal of Medicine. 2020;382(7):622–631.
- Heerspink HJL, Stefansson BV, Correa-Rotter R, et al. Dapagliflozin in patients with chronic kidney disease (DAPA-CKD). New England Journal of Medicine. 2020;383(15):1436–1446.
- Roberts ISD. Oxford Classification of IgA nephropathy: an update. Kidney International Supplements. 2018;8(1):5–7.
Research Papers
Explore current literature on glomerulonephritis via PubMed topic searches. These links open live PubMed searches for the listed keywords — results update as new studies are indexed.
- Clinical trials on glomerulonephritis — PubMed search
- Clinical trials on IgA nephropathy — PubMed search
- Clinical trials on ANCA-associated vasculitis — PubMed search
- Clinical trials on anti-GBM disease Goodpasture — PubMed search
- Clinical trials on lupus nephritis — PubMed search
- Clinical trials on membranous nephropathy — PubMed search
- Clinical trials on rapidly progressive glomerulonephritis — PubMed search
Connections
- Nephrology & Hepatology
- Nephrotic Syndrome
- Kidney Disease
- Lupus
- Hypertension
- Diabetes
- Hepatitis C
- Acute Kidney Injury
- eGFR
- Creatinine
- Edema
- Hepatitis B
- Urinalysis
- Kidney Function Tests
- Cirrhosis
- Cardiovascular Disease
- Lymphoma
- Sjogrens Syndrome
- Lupus Nephritis and Kidney Involvement
- Strep Throat
- Polycystic Kidney Disease
- Vasculitis — the systemic vessel inflammation whose ANCA-associated forms produce pauci-immune crescentic GN.