Thalassemia
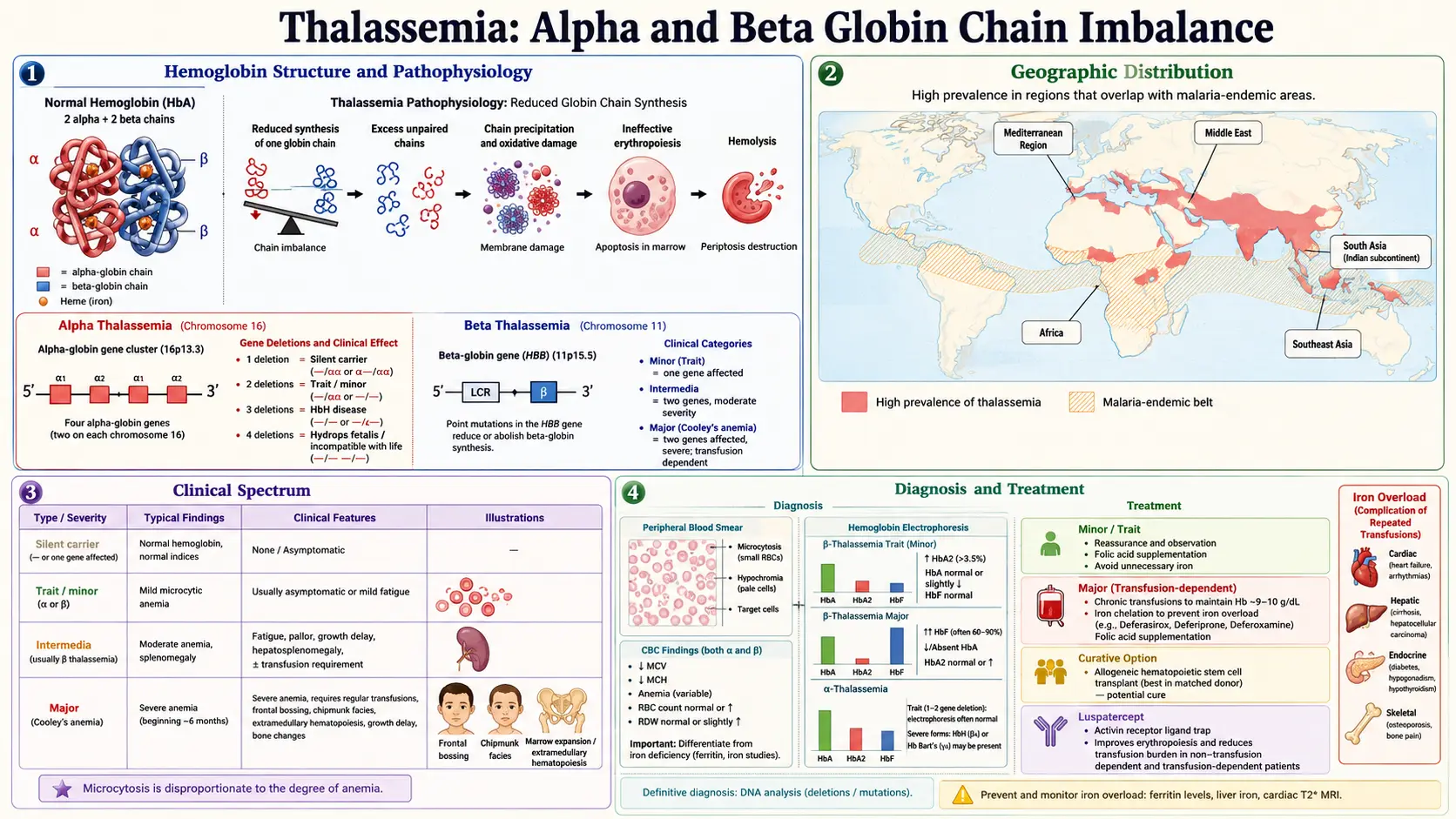
Table of Contents
- Table of Contents
- What Is Thalassemia?
- Alpha vs Beta Thalassemia
- Classification by Severity
- Genetics and Inheritance Patterns
- Geographic Prevalence
- Symptoms by Severity
- Diagnosis
- Conventional Treatment
- Natural Support Strategies
- Iron Overload from Transfusions
- Nutrition Guidelines
- Psychological Support
- Genetic Counseling
- Important Cautions
- Research Papers
- Connections
- Featured Videos
What Is Thalassemia?
Thalassemia is a group of inherited blood disorders characterized by the body's inability to produce adequate amounts of hemoglobin, the oxygen-carrying protein in red blood cells. The name derives from the Greek word "thalassa" (sea), as the condition was first identified in populations around the Mediterranean Sea.
Hemoglobin consists of two types of protein chains: alpha-globin and beta-globin. Normal adult hemoglobin (HbA) contains two alpha chains and two beta chains. In thalassemia, mutations in the genes responsible for producing one or both of these chains result in reduced or absent production, leading to defective hemoglobin molecules, ineffective red blood cell production, and chronic anemia.
From a naturopathic perspective, thalassemia presents unique challenges because the anemia cannot be corrected by nutritional interventions alone. However, natural approaches play a vital supportive role in managing complications, optimizing nutrition, reducing oxidative stress, and improving quality of life.
Alpha vs Beta Thalassemia
Alpha Thalassemia
Alpha-globin is encoded by four genes (two on each copy of chromosome 16). The severity of alpha thalassemia depends on how many of these four genes are affected:
- Silent carrier (1 gene affected) — No symptoms, normal blood counts, detectable only by genetic testing
- Alpha thalassemia trait (2 genes affected) — Mild microcytic anemia, often mistaken for iron deficiency
- Hemoglobin H disease (3 genes affected) — Moderate to severe anemia, splenomegaly, jaundice, and bone changes. Hemoglobin H (beta-4 tetramers) forms and is unstable.
- Hydrops fetalis / Bart's hemoglobin (4 genes affected) — Incompatible with life. Affected fetuses typically die in utero or shortly after birth due to severe anemia and heart failure.
Beta Thalassemia
Beta-globin is encoded by two genes (one on each copy of chromosome 11). Mutations can either reduce (beta-plus) or completely eliminate (beta-zero) beta-globin production:
- Beta thalassemia minor / trait (1 gene affected) — Mild anemia, usually asymptomatic
- Beta thalassemia intermedia — Moderate anemia that may or may not require transfusions
- Beta thalassemia major (Cooley's anemia) — Severe anemia requiring lifelong regular blood transfusions, typically beginning in infancy
Classification by Severity
Thalassemia Trait (Minor)
Individuals with thalassemia trait carry one normal and one affected gene. They are generally healthy with mild anemia characterized by small red blood cells (low MCV) and a slightly reduced hemoglobin level. Most people with thalassemia trait are unaware of their status until routine blood work reveals the characteristic microcytic pattern. No treatment is typically required, though awareness is important for family planning.
Thalassemia Intermedia
This represents a moderate form where patients have significant anemia but may not require regular transfusions. Symptoms can include moderate fatigue, enlarged spleen, bone deformities, and gallstones. Some patients may need occasional transfusions during illness, pregnancy, or surgery. The clinical picture varies widely, and management is individualized.
Thalassemia Major (Cooley's Anemia)
The most severe form, typically diagnosed in the first two years of life when fetal hemoglobin (HbF) is replaced by defective adult hemoglobin. Without treatment, thalassemia major causes:
- Severe anemia with hemoglobin levels as low as 3-4 g/dL
- Failure to thrive, poor growth, and developmental delays
- Massive bone marrow expansion causing characteristic facial bone changes (frontal bossing, prominent cheekbones)
- Massive hepatosplenomegaly (enlarged liver and spleen)
- Death in early childhood without intervention
With modern transfusion therapy and iron chelation, patients with thalassemia major can survive well into adulthood, though lifelong medical management is required.
Genetics and Inheritance Patterns
Thalassemia follows an autosomal recessive inheritance pattern. This means:
- A child must inherit a defective gene from both parents to develop thalassemia major or intermedia.
- If both parents carry thalassemia trait, each pregnancy carries a 25% chance of thalassemia major, a 50% chance of thalassemia trait, and a 25% chance of being unaffected.
- Carriers (thalassemia trait) can pass the gene to their children but typically do not have significant symptoms themselves.
Alpha thalassemia genetics are more complex because four genes are involved. The pattern of gene deletion (whether on the same chromosome or different chromosomes) affects the risk of having severely affected children. The cis configuration (both deletions on the same chromosome), more common in Southeast Asian populations, carries a higher risk of hydrops fetalis in offspring.
Geographic Prevalence
Thalassemia is most prevalent in populations from regions where malaria has been historically endemic. Carrying thalassemia trait provides a survival advantage against malaria, which explains the high carrier rates in certain geographic regions:
- Mediterranean region — Italy (particularly Sardinia and Sicily), Greece, Turkey, and Cyprus have beta thalassemia carrier rates of 5-20%
- Southeast Asia — Thailand, Cambodia, Laos, Vietnam, Malaysia, Indonesia, and the Philippines have high rates of both alpha and beta thalassemia. Alpha thalassemia carrier rates exceed 30% in some areas.
- Middle East — Iran, Iraq, Saudi Arabia, and other Gulf states have significant beta thalassemia prevalence
- South Asia — India, Pakistan, Bangladesh, and Sri Lanka have substantial carrier populations
- Africa — Alpha thalassemia is particularly common across sub-Saharan Africa, with carrier rates of 20-30% in some regions
- China — Both alpha and beta thalassemia are common in southern China
With global migration, thalassemia is now found in populations worldwide, including North America, Europe, and Australia.
Symptoms by Severity
Thalassemia Trait (Mild)
- Usually asymptomatic
- Mild fatigue in some individuals
- Slightly pale appearance
- Low MCV on blood count (often mistaken for iron deficiency)
Thalassemia Intermedia (Moderate)
- Moderate fatigue and reduced exercise tolerance
- Pallor
- Enlarged spleen (splenomegaly)
- Jaundice (yellowish skin and eyes from red blood cell destruction)
- Gallstones (pigmented, from chronic hemolysis)
- Bone changes — Mild facial bone expansion, osteoporosis
- Leg ulcers
- Iron overload (even without transfusions, due to increased intestinal absorption)
Thalassemia Major (Severe)
- Severe anemia appearing in the first 6-24 months of life
- Failure to thrive — Poor growth, feeding difficulties
- Severe pallor and jaundice
- Massive splenomegaly and hepatomegaly
- Characteristic facial changes — Frontal bossing, prominent cheekbones, dental malocclusion from bone marrow expansion
- Skeletal deformities — Thinning of long bones, pathological fractures
- Delayed puberty and growth retardation
- Iron overload complications (from chronic transfusions)
Diagnosis
Complete Blood Count (CBC)
- Low hemoglobin — Varies from mildly reduced (trait) to severely low (major)
- Low MCV (mean corpuscular volume) — Characteristically small red blood cells
- Low MCH (mean corpuscular hemoglobin) — Reduced hemoglobin per cell
- Elevated red cell distribution width (RDW) — Variable red blood cell sizes
- Peripheral blood smear — Shows target cells, hypochromic microcytic cells, and nucleated red blood cells in severe cases
Hemoglobin Electrophoresis
This is the key diagnostic test that separates and quantifies the different types of hemoglobin:
- Beta thalassemia trait — Elevated HbA2 (above 3.5%) and possibly elevated HbF
- Beta thalassemia major — Very high HbF (60-90%), elevated HbA2, reduced or absent HbA
- Alpha thalassemia — May show Hemoglobin H (HbH) or Bart's hemoglobin in more severe forms. Mild alpha thalassemia may have a normal electrophoresis pattern.
Genetic Testing
- DNA analysis — Identifies specific mutations in alpha or beta-globin genes
- Essential for alpha thalassemia diagnosis (which may not be apparent on electrophoresis)
- Important for genetic counseling and prenatal diagnosis
Iron Studies
- Serum ferritin and transferrin saturation — To assess iron status and distinguish from iron deficiency
- Important because thalassemia trait is frequently misdiagnosed as iron deficiency, leading to inappropriate iron supplementation
Conventional Treatment
Blood Transfusions
Regular blood transfusions are the cornerstone of treatment for thalassemia major:
- Transfusions every 2-4 weeks to maintain pre-transfusion hemoglobin above 9-10 g/dL
- Suppresses ineffective erythropoiesis and bone marrow expansion
- Allows normal growth and development in children
- Reduces complications of chronic anemia
- Risks include iron overload, alloimmunization, and transfusion reactions
Iron Chelation Therapy
Essential for all regularly transfused patients to prevent fatal iron overload:
- Deferoxamine (Desferal) — Subcutaneous infusion over 8-12 hours, typically 5-7 nights per week. Highly effective but burdensome.
- Deferasirox (Exjade/Jadenu) — Once-daily oral tablet. More convenient, now the most widely used chelator.
- Deferiprone (Ferriprox) — Oral chelator taken three times daily. Particularly effective for cardiac iron removal and often used in combination with deferoxamine.
Bone Marrow Transplant (BMT)
Allogeneic hematopoietic stem cell transplantation is currently the only curative treatment for thalassemia major:
- Best outcomes when performed early in childhood from an HLA-matched sibling donor
- Success rates exceed 80-90% with matched sibling donors in well-selected patients
- Risks include graft-versus-host disease, graft failure, and transplant-related mortality
- Limited by donor availability — only about 25% of patients have a matched sibling
Gene Therapy Research
Gene therapy represents an exciting frontier in thalassemia treatment:
- Betibeglogene autotemcel (Zynteglo) — The first gene therapy approved for transfusion-dependent beta thalassemia. Uses a lentiviral vector to insert a functional beta-globin gene into the patient's own stem cells.
- CRISPR-based therapies — Emerging approaches that edit genes to reactivate fetal hemoglobin production, effectively compensating for the defective beta-globin.
- Gene therapy offers the possibility of a cure without the risks of donor transplantation.
Splenectomy
Removal of the spleen may be considered when:
- Transfusion requirements increase significantly due to hypersplenism
- The spleen causes mechanical discomfort due to massive enlargement
- Risks include increased susceptibility to encapsulated bacterial infections — prophylactic antibiotics are essential post-splenectomy
Natural Support Strategies
Natural approaches in thalassemia focus on reducing oxidative stress, supporting organ function, and optimizing nutritional status. These are supportive measures that complement conventional treatment.
Folic Acid
Folic acid (vitamin B9) is essential for red blood cell production and is rapidly depleted in thalassemia due to the bone marrow's increased demand for new red blood cells. Supplementation of 1-5 mg daily is widely recommended for thalassemia intermedia and major patients, particularly those not on regular transfusions.
Vitamin E
Vitamin E is a fat-soluble antioxidant that protects red blood cell membranes from oxidative damage. Thalassemia patients typically have low vitamin E levels due to increased oxidative stress. Studies have shown that vitamin E supplementation can reduce red blood cell destruction and may modestly improve hemoglobin levels. Doses of 400-800 IU daily are commonly used under medical supervision.
Antioxidant Support
The chronic hemolysis and iron overload in thalassemia generate significant oxidative stress. Antioxidant support may include:
- N-acetyl cysteine (NAC) — Supports glutathione production, the body's master antioxidant. May reduce oxidative damage to red blood cells.
- Curcumin — Has iron-chelating and antioxidant properties. Some studies suggest it may help reduce iron burden and protect the liver.
- Green tea extract (EGCG) — Potent antioxidant with mild iron-chelating activity
- Silymarin (milk thistle) — Hepatoprotective antioxidant that may support liver health in iron-overloaded patients
L-Carnitine
L-carnitine plays a critical role in cellular energy metabolism by transporting fatty acids into mitochondria. Thalassemia patients frequently have carnitine deficiency due to increased energy demands and oxidative stress. Supplementation (1-2 g daily) has been shown to improve fatigue, cardiac function, and exercise tolerance in some studies.
Vitamin D
Vitamin D deficiency is extremely common in thalassemia patients (prevalence of 60-80%), contributing to osteoporosis, bone pain, and increased fracture risk. Factors contributing to deficiency include iron overload affecting vitamin D metabolism, liver dysfunction, and reduced sun exposure. Supplementation should be guided by serum 25-hydroxyvitamin D levels, with a target of 30-50 ng/mL.
Zinc
Zinc deficiency is common in thalassemia, partly because iron chelation therapy (particularly deferoxamine) can deplete zinc. Zinc is important for growth, immune function, and wound healing. Supplementation may be beneficial, especially in children with growth retardation.
Iron Overload from Transfusions
Iron overload is the most serious complication of chronic transfusion therapy and the leading cause of morbidity and mortality in well-transfused thalassemia patients. Each unit of transfused blood contains approximately 200-250 mg of iron, and the body has no mechanism to actively excrete this excess.
Cardiac Iron Overload
- The leading cause of death in thalassemia major
- Iron deposits in cardiac muscle cause cardiomyopathy, arrhythmias, and heart failure
- Monitored by cardiac MRI T2* — values below 20 milliseconds indicate cardiac iron loading, below 10 ms indicates severe risk
- Cardiac iron overload can be reversed with intensive chelation therapy, particularly using deferiprone alone or in combination with deferoxamine
Liver Iron Overload
- Iron accumulates first in the liver, causing fibrosis and eventually cirrhosis
- Monitored by liver MRI (R2 or T2*) or liver iron concentration (LIC)
- Target LIC is below 7 mg/g dry weight; above 15 mg/g indicates severe overload with high risk of cardiac involvement
- Hepatitis C co-infection (from historical transfusion practices) accelerates liver damage
Endocrine Complications
- Diabetes mellitus — From iron damage to the pancreas
- Hypogonadism — Delayed puberty, infertility, reduced bone density
- Hypothyroidism
- Hypoparathyroidism — Leading to hypocalcemia
- Growth hormone deficiency — Contributing to short stature in children
Nutrition Guidelines
Nutrition management in thalassemia must balance the need for adequate nutrients with the avoidance of excess iron:
General Recommendations
- Avoid iron supplements and iron-fortified foods (unless iron deficiency is documented, which is rare in transfused patients)
- Consume tea or coffee with meals to reduce non-heme iron absorption
- Include calcium-rich foods with meals to inhibit iron absorption
- Emphasize whole grains and legumes — Their phytate content naturally limits iron absorption
- Limit red meat — Contains highly bioavailable heme iron
- Moderate vitamin C intake with meals — Avoid large doses with food as it enhances iron absorption
Key Nutrients to Optimize
- Calcium — Essential for bone health (1,000-1,500 mg daily), especially given the high risk of osteoporosis
- Vitamin D — Critical for calcium absorption and bone mineralization
- Folic acid — Supports red blood cell production
- Vitamin E — Antioxidant protection for red blood cells
- Zinc — Often depleted by chelation therapy
- B vitamins — Support energy metabolism and overall health
Psychological Support
Living with thalassemia presents significant psychological and social challenges that are often underappreciated:
- Chronic illness burden — Regular hospital visits for transfusions and monitoring, daily chelation therapy, and the knowledge of a lifelong condition create ongoing stress.
- Body image concerns — Facial bone changes, short stature, delayed puberty, and skin changes from iron overload can affect self-esteem, particularly during adolescence.
- Anxiety and depression — Studies show higher rates of anxiety and depression in thalassemia patients compared to the general population. Regular mental health screening is recommended.
- Treatment adherence — Chelation therapy compliance is a major challenge, particularly in adolescents. Non-adherence leads to iron overload complications.
- Transition to adult care — Moving from pediatric to adult services is a vulnerable period requiring careful planning and support.
- Peer support — Connecting with thalassemia support organizations and peer groups can significantly improve coping and quality of life.
- Mindfulness and stress reduction — Meditation, yoga, and other mind-body practices can help manage the emotional burden of chronic disease.
Genetic Counseling
Genetic counseling is a cornerstone of thalassemia prevention and management:
- Carrier screening — Recommended for all individuals from high-prevalence populations, ideally before starting a family. A simple CBC and hemoglobin electrophoresis can identify most carriers.
- Partner testing — If one partner is a carrier, the other should be tested. If both are carriers, genetic counseling should outline the reproductive risks and options.
- Prenatal diagnosis — Chorionic villus sampling (CVS) at 10-12 weeks or amniocentesis at 15-18 weeks can determine if a fetus is affected. Preimplantation genetic diagnosis (PGD) with IVF is also an option.
- Population screening programs — Countries like Cyprus, Sardinia, Greece, and Iran have implemented successful screening programs that have dramatically reduced the birth rate of thalassemia major.
- Emotional support — Genetic counseling should include emotional and psychological support for families facing difficult reproductive decisions.
Important Cautions
- Do not take iron supplements unless specifically directed by a hematologist. Thalassemia trait is frequently misdiagnosed as iron deficiency anemia, leading to harmful iron supplementation. A proper diagnosis with hemoglobin electrophoresis is essential before any iron therapy.
- Natural supplements cannot replace transfusion therapy in thalassemia major. Failure to maintain adequate hemoglobin through transfusions leads to severe complications and death.
- Iron chelation therapy is life-saving and must not be discontinued or replaced with natural chelators alone. While compounds like curcumin and IP6 may offer modest supportive benefit, they are not potent enough to manage transfusion-related iron overload.
- Folic acid supplementation should be discussed with your hematologist, as it may not be necessary in well-transfused patients whose bone marrow activity is suppressed.
- Avoid oxidant drugs — Certain medications (such as some antibiotics and antimalarials) can worsen hemolysis in thalassemia. Always inform healthcare providers of your diagnosis.
- Splenectomy increases infection risk — Post-splenectomy patients may require lifelong prophylactic antibiotics. Any fever should be treated as a medical emergency.
- Pregnancy in thalassemia requires specialized obstetric care due to risks of worsening anemia, cardiac complications, and the need for adjusted transfusion and chelation protocols.
- Consult a hematologist experienced in thalassemia management before adding any natural supplements, as interactions with chelation therapy and other medications are possible.
Research Papers
The following PubMed topic searches return current peer-reviewed literature relevant to this condition. Each link opens a live PubMed query.
- Beta thalassemia
- Alpha thalassemia
- Thalassemia major transfusion
- Iron chelation thalassemia
- Luspatercept thalassemia
- Thalassemia hematopoietic stem cell transplant
- Thalassemia gene therapy
- Thalassemia intermedia
- Thalassemia prenatal screening
- Hemoglobinopathy genetics
- Thalassemia management guidelines
- Thalassemia cardiac iron overload
Connections
- Hematology
- Anemia
- Iron
- Sickle Cell Disease
- Hemochromatosis
- Heart Failure
- Cardiomyopathy
- Folate
- Complete Blood Count
- Osteoporosis
- Zinc
- Vitamin E
- Fatigue
- Jaundice
- Malaria
- Calcium
- NAC
- Iron Deficiency Anemia
- Hemophilia