Thiamine for Cognitive Function and Alzheimer's Disease
Thiamine deficiency in mild cognitive impairment (MCI) and early Alzheimer's disease is a distinct, separate phenomenon from the full Korsakoff amnesia of acute thiamine collapse. Brain FDG-PET in early AD shows cerebral glucose hypometabolism in the same temporal-parietal regions where postmortem studies show reduced activity of the two thiamine-dependent enzymes pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase (α-KGDH). This pattern preceeds amyloid deposition by years — cerebral energy failure may be a primary driver, not just a downstream consequence. Gibson's 2020 benfotiamine 1-year pilot trial in mild AD produced encouraging signals on the ADAS-Cog and MMSE, and a larger phase 2 trial (NCT02292238) is underway. This page covers the cerebral-glucose-hypometabolism story, the Gibson trial and its predecessor work, the distinction between subclinical thiamine-related cognitive decline and full Wernicke-Korsakoff, the alcohol-related-cognitive-impairment overlap, and why thiamine + mitochondrial support sits on the same reading list as alpha lipoic acid for cognitive aging.
Table of Contents
- The Thiamine-Cognition Link
- Cerebral Glucose Hypometabolism in Early AD
- PDH and α-KGDH Activity in Postmortem AD Brain
- Thiamine Status in Mild Cognitive Impairment
- The Gibson 2020 Benfotiamine Pilot Trial
- Mechanistic Pathway: From Low TPP to Cognitive Decline
- Alcohol-Related Cognitive Impairment vs Korsakoff
- How This Differs from Wernicke-Korsakoff
- Thiamine + ALA for Cognitive Aging
- Practical Protocol for Cognitive Concerns
- Cautions
- Key Research Papers
- Connections
- Featured Videos
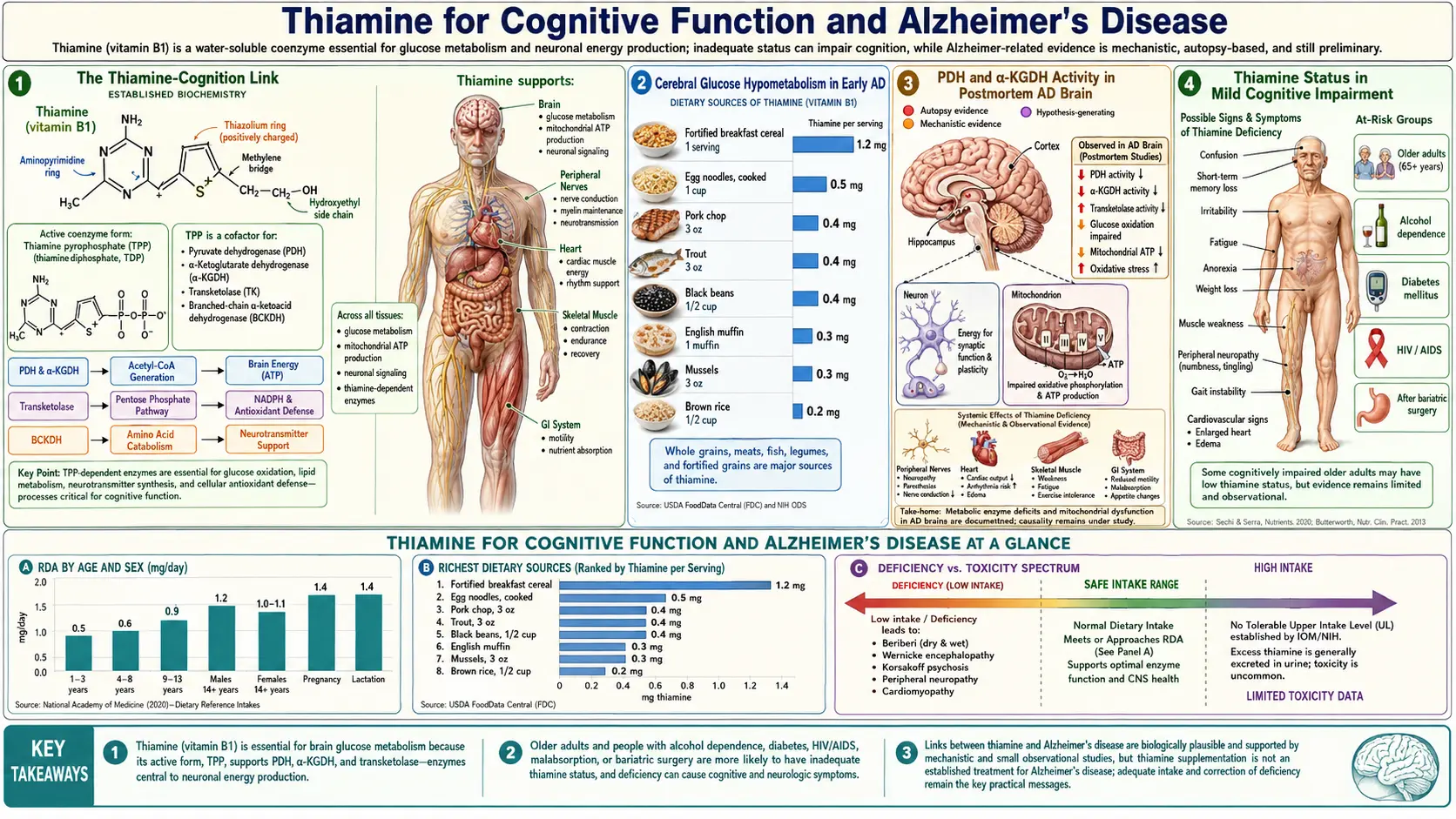
The Thiamine-Cognition Link
The connection between thiamine and brain function was first established a century ago through the dramatic neuropsychiatric manifestations of severe deficiency — Wernicke encephalopathy and Korsakoff amnestic syndrome. But starting in the 1980s, a separate, more subtle, more common picture began to emerge: subclinical thiamine deficiency contributes to age-related cognitive decline and may play a meaningful role in the early phases of Alzheimer's disease.
This is not the same disease as Korsakoff syndrome. Korsakoff syndrome is the catastrophic dementia that follows acute neurological emergency — it presents abruptly, dramatically, and produces a profound permanent anterograde amnesia. The thiamine-cognition link described here is gradual, subclinical, and clinically resembles the early phases of "normal" aging or mild Alzheimer's disease. The biochemistry is the same (impaired TPP-dependent enzymes) but the dose and timing are different — chronic mild deficiency over years rather than acute severe collapse over weeks.
Three converging lines of evidence support the link:
- FDG-PET imaging — the same temporal-parietal regions that lose glucose uptake in early AD are TPP-enzyme-dependent regions
- Postmortem brain enzyme studies — PDH and α-KGDH activity is reduced in AD-affected brain regions, even when the thiamine concentration appears adequate
- Pilot intervention trials — benfotiamine has shown promising signals in MCI and early AD, with a larger trial underway
The unifying framework: cerebral energy failure (a "brain-as-a-glucose-engine-running-out-of-fuel" story) may be a primary upstream driver of Alzheimer's disease, and thiamine repletion through high-bioavailability forms (benfotiamine, allithiamine) is a candidate disease-modifying intervention. This is in active debate but the supporting evidence base has grown substantially over the past decade.
Cerebral Glucose Hypometabolism in Early AD
FDG-PET (fluorodeoxyglucose positron emission tomography) measures cerebral glucose uptake region by region. In healthy older adults, glucose uptake is roughly uniform across cortical regions. In Alzheimer's disease, distinctive regional patterns of reduced glucose uptake appear, often years before clinical dementia is detectable:
- Posterior cingulate and precuneus — among the earliest regions affected; hypometabolism here can predate clinical AD by 10-15 years in some studies
- Lateral temporal cortex — particularly the temporo-parietal junction
- Parietal association cortex — bilateral, often symmetric
- Lateral prefrontal cortex — later in disease progression
- Relative sparing of primary sensorimotor cortex, occipital cortex, and basal ganglia until late in disease
The pattern is so distinctive that FDG-PET is used clinically to distinguish AD from other dementias (frontotemporal dementia, Lewy body dementia have different regional patterns). Crucially, the hypometabolism appears before significant amyloid plaque deposition becomes visible on amyloid PET, and before clinical symptoms become detectable. This temporal precedence raises the question: is impaired glucose metabolism a primary driver of AD, or merely a downstream consequence of amyloid pathology?
The thiamine-cognition framework leans toward the primary-driver interpretation: if TPP-dependent enzymes lose activity, glucose entering the brain cannot be efficiently metabolized to ATP, regional ATP supply falls, and downstream pathology (synaptic dysfunction, neuronal stress, eventual amyloid and tau deposition) follows. Repleting TPP would, on this model, address the upstream metabolic failure rather than the downstream protein aggregates.
This view is consistent with the otherwise puzzling repeated failure of amyloid-targeting drugs to produce robust clinical benefit in AD trials — if amyloid is downstream of metabolic failure, removing amyloid without addressing the upstream metabolism would predict modest at best clinical benefit, which is exactly what has been observed.
PDH and α-KGDH Activity in Postmortem AD Brain
The pioneering postmortem brain enzyme studies of Gary Gibson and colleagues at the Weill Cornell Burke Neurological Institute documented across several decades of work that:
- α-KGDH activity is reduced by 50-75% in the cortex of patients dying with Alzheimer's disease compared to age-matched controls
- PDH activity is reduced, though to a smaller degree, in the same brain regions
- The third TPP-dependent enzyme, transketolase, is also reduced
- The reductions are most pronounced in the brain regions that show the FDG-PET hypometabolism pattern
- The reductions are present even when measured TPP concentration in the brain tissue appears within the normal range — suggesting that something beyond simple TPP availability is impairing the enzymes
- The reductions appear early in disease, not just in end-stage tissue
Several mechanisms have been proposed for why these enzymes lose activity in AD even with adequate TPP:
- Oxidative damage to the enzyme proteins themselves — AD brain has elevated markers of oxidative and nitrosative stress, and the lipoamide-dependent enzymes (PDH, α-KGDH, BCKDH) are particularly susceptible to oxidative inactivation
- Reduced lipoylation — the lipoamide cofactor of these enzymes is depleted in AD brain (interestingly, this is also where alpha lipoic acid mechanism stories intersect)
- Reduced expression of enzyme subunits — transcriptional changes in AD brain reduce production of the enzyme proteins
- Increased demand for repair — functional thiamine deficiency despite normal apparent concentration
The clinical relevance: even patients with normal serum thiamine and normal-appearing brain TPP concentration may benefit from supranormal TPP levels (achievable with benfotiamine or other high-bioavailability forms) by forcing more substrate through the partially-inactivated enzymes. This is the mechanistic rationale for the Gibson trial.
Thiamine Status in Mild Cognitive Impairment
Several cross-sectional and observational studies have examined thiamine status in patients with mild cognitive impairment (MCI):
- Erythrocyte transketolase activity (the functional thiamine marker) is reduced in patients with MCI compared to age-matched cognitively normal controls in multiple cohorts
- The activation coefficient (the in vitro response to TPP loading) is elevated in MCI, indicating functional thiamine deficiency
- Whole-blood TPP measured by HPLC shows lower values in MCI patients
- The thiamine-status reduction in MCI is modest in magnitude but consistent in direction across studies
- The thiamine reduction correlates with cognitive scores and with the FDG-PET hypometabolism pattern
It is not yet clear whether thiamine deficiency causes the MCI or whether MCI (with its associated changes in diet, appetite, mood, and brain biochemistry) causes the thiamine deficiency. Both directions are plausible and both are probably operating — a vicious cycle in which subclinical thiamine deficiency drives cognitive decline, which then worsens dietary intake and further reduces thiamine status.
The pragmatic implication for clinicians and patients: thiamine status should be considered and optimized in any patient with MCI or early AD. The cost of supplementation is trivial, the safety profile is excellent, and even if the disease-modifying potential turns out to be modest, ensuring adequate thiamine status is straightforward general supportive care.
The Gibson 2020 Benfotiamine Pilot Trial
The Gibson et al. 2020 study (published in Journal of Alzheimer's Disease) was a randomized, double-blind, placebo-controlled pilot trial of benfotiamine 600 mg/day vs placebo for 12 months in 70 patients with amnestic MCI or mild AD. The primary outcomes were change in the ADAS-Cog (Alzheimer's Disease Assessment Scale - Cognitive subscale) and the Clinical Dementia Rating Sum of Boxes (CDR-SB).
Results:
- Benfotiamine 600 mg/day was well-tolerated; no significant adverse events distinguished it from placebo
- Numerical improvement (smaller decline) in cognitive scores in the benfotiamine group, with a trend toward statistical significance in the per-protocol analysis
- The treatment effect was larger in patients without the APOEε4 risk allele — suggesting benfotiamine may be more effective in non-genetic AD, or that APOEε4 carriers may need higher doses
- Blood thiamine markers confirmed compliance and adequate absorption
- FDG-PET showed evidence of stabilized cerebral glucose metabolism in the benfotiamine group
- The study was underpowered to detect a definitive effect but produced sufficient signal to justify a larger phase 2 trial
A larger trial — the Burke Foundation BenfoTeam phase 2 study (NCT02292238 and successors) — is now underway to definitively test whether benfotiamine slows cognitive decline in mild AD. Results are pending but if positive would represent the first nutritionally-based disease-modifying intervention in AD.
The Gibson trial is an example of how mechanistic and biochemical evidence can drive a rational therapeutic candidate even in the absence of a definitive large trial. Many integrative-medicine and functional-medicine practitioners now recommend benfotiamine 300-600 mg/day for patients with MCI or early AD, based on the favorable risk-benefit balance even pending definitive trial data.
Mechanistic Pathway: From Low TPP to Cognitive Decline
The proposed mechanistic chain from low TPP to cognitive decline runs as follows:
- Reduced intracellular TPP — from inadequate dietary intake, age-related malabsorption, alcohol use, medications that interfere with absorption, or oxidative damage to TPP itself
- Reduced PDH activity — pyruvate cannot efficiently enter the Krebs cycle, glucose is shunted to lactate, cerebral ATP supply drops
- Reduced α-KGDH activity — the rate-limiting Krebs cycle enzyme loses activity, further collapsing ATP production
- Reduced transketolase activity — the pentose phosphate pathway loses flux, generating less NADPH for glutathione recycling and antioxidant defense
- Cerebral energy supply falls — particularly in the temporo-parietal regions that have the highest energy demand and are most vulnerable
- Synaptic dysfunction begins — ATP-dependent vesicle recycling, ion pumping, and neurotransmitter synthesis all suffer
- Oxidative stress rises — both because ATP supply for antioxidant systems falls and because the impaired mitochondria leak more superoxide
- Glutamate excitotoxicity engages — ATP-dependent glutamate uptake fails, NMDA receptors fire excessively, calcium floods into neurons
- Cognitive symptoms emerge — first as MCI, then as mild AD, eventually as moderate-to-severe dementia
- Pathologic protein deposition (amyloid, tau) accumulates — possibly secondary to the cumulative metabolic and oxidative stress, possibly through impaired clearance of misfolded proteins
Benfotiamine intervenes early in this chain by driving up intracellular TPP and reactivating the partially-inhibited enzymes. The hope is that this slows or arrests the cascade before it produces irreversible neuronal loss.
This framework also explains why pairing benfotiamine with mitochondrial support like alpha lipoic acid is rational — ALA addresses the downstream oxidative-stress and lipoamide-cofactor parts of the same biochemistry that benfotiamine addresses upstream. The combination is more comprehensive than either alone.
Alcohol-Related Cognitive Impairment vs Korsakoff
Chronic alcohol use produces a spectrum of cognitive consequences that includes:
- Subtle executive dysfunction — impaired attention, working memory, decision-making; reversible with abstinence in the early phases
- Mild alcohol-related cognitive impairment — more pronounced but still partially reversible; persists despite abstinence in moderate cases
- Alcohol-related brain damage / alcohol-related dementia — permanent cognitive impairment from cumulative neurotoxicity, frontal lobe atrophy on MRI, often partially overlapping with Korsakoff features but not requiring a documented Wernicke episode
- Wernicke-Korsakoff syndrome — the acute-then-chronic catastrophic dementia following acute thiamine collapse, classically with the Wernicke triad followed by Korsakoff amnesia
The first three entities all have a substantial thiamine-deficiency component on top of direct alcohol neurotoxicity. Alcohol impairs thiamine absorption, accelerates urinary loss, depletes liver stores, and substitutes empty calories for thiamine-rich food. Most heavy drinkers have low to borderline thiamine status, and the contribution of thiamine deficiency to alcohol-related cognitive decline is hard to disentangle from direct alcohol neurotoxicity.
Pragmatically: any patient with significant alcohol use history and cognitive concerns should receive long-term thiamine supplementation (oral thiamine HCl 100 mg/day or benfotiamine 300 mg/day), particularly during and after abstinence. Some recovery of cognitive function is possible with sustained abstinence + thiamine repletion, even years into chronic alcohol-related cognitive impairment. Continued drinking after thiamine repletion typically does not preserve cognitive function — the direct alcohol neurotoxicity continues regardless.
It is worth noting that alcohol-related cognitive impairment is distinct from Korsakoff syndrome in important ways. Korsakoff is characterized by profound anterograde amnesia, confabulation, and mammillary body atrophy on MRI — a relatively focal deficit. Alcohol-related dementia tends to be more global, with prominent executive dysfunction, frontal atrophy on MRI, and impairment across multiple cognitive domains. The two can coexist in the same patient.
How This Differs from Wernicke-Korsakoff
| Feature | Wernicke-Korsakoff | Subclinical Thiamine-Cognition Link |
|---|---|---|
| Onset | Acute, days to weeks | Gradual, months to years |
| Severity of deficiency | Severe, often total store depletion | Mild to moderate, often "low normal" |
| Clinical phenotype | Triad: ophthalmoplegia + ataxia + confusion → profound anterograde amnesia | Resembles MCI / early AD: episodic memory, executive function, processing speed |
| Imaging | Mammillary body atrophy, thalamic atrophy | FDG-PET hypometabolism in temporo-parietal regions |
| Treatment | Parenteral thiamine 500 mg TID acute, then maintenance | Oral benfotiamine 300-600 mg/day chronically; oral thiamine HCl 100 mg/day adequate for prevention |
| Reversibility | Partial; depends on speed of treatment | Possibly modest; mechanism is "slow the decline" rather than "reverse the deficit" |
| Mortality without treatment | ~17-20% | Not directly fatal; contributes to broader cognitive decline mortality |
| Diagnostic approach | Clinical recognition + empirical thiamine; MRI confirms | Standard cognitive testing + thiamine status (erythrocyte transketolase, whole-blood TPP) |
Both conditions reflect the same biochemistry — impaired TPP-dependent enzyme function — but at very different doses and time scales. Severe acute deficiency produces the Wernicke-Korsakoff phenotype. Chronic mild deficiency over years produces the gradual cognitive decline that resembles aging or early AD. Recognizing the latter requires a different clinical lens than recognizing the former.
Thiamine + ALA for Cognitive Aging
Thiamine and alpha lipoic acid are mechanistically tightly linked in cognitive aging:
- Both target the same enzymes — PDH and α-KGDH both require TPP and lipoamide as cofactors. TPP catalyzes the decarboxylation step; lipoamide (covalently bound to the E2 subunit) carries the acyl group to coenzyme A. If either cofactor is depleted, the enzyme fails. Thiamine repletion drives TPP; lipoic acid drives lipoamide. Together they restore both ends of the cofactor pair.
- Both cross the blood-brain barrier — benfotiamine, allithiamine, and ALA all enter the brain readily, where they can directly support neuronal energy production.
- Both have favorable safety profiles — the combination has been used in integrative cognitive-aging protocols for decades without significant adverse interaction.
- Both can be combined with acetyl-L-carnitine — the Ames-Hagen group at UC Berkeley demonstrated that the ALA + acetyl-L-carnitine combination reversed mitochondrial decline in aged rat brain. Adding benfotiamine to this stack provides the missing TPP support.
- The combination is supported in multiple cognitive-aging protocols — though no head-to-head trial of the triple combination (benfotiamine + ALA + ALCAR) has been done, the mechanism is sound and the components have individual evidence bases.
A typical cognitive-support stack: benfotiamine 300 mg twice daily + alpha lipoic acid 600 mg once daily + acetyl-L-carnitine 1-2 g/day + a comprehensive B-complex + magnesium 200-400 mg/day. The combination targets multiple mechanisms simultaneously and has a favorable safety profile, though the empirical evidence for the combination specifically is more about mechanistic plausibility than head-to-head trial data.
This is not a treatment for established moderate-to-severe AD — the evidence is not strong enough for that claim, and the conventional anti-cholinesterase medications (donepezil, rivastigmine, galantamine) and the newer amyloid-targeting antibodies remain the standard of care. But for patients with MCI, early AD, or cognitive aging concerns, the benfotiamine + ALA combination is a reasonable adjunct with mechanism, safety, and emerging trial support.
Practical Protocol for Cognitive Concerns
For patients with MCI, early AD, alcohol-related cognitive concerns, or general cognitive-aging interest:
Tier 1 (everyone)
- Thiamine HCl 100 mg/day orally, or benfotiamine 300 mg/day for higher bioavailability
- Comprehensive B-complex — ensures adequate B2, B3, B6, B12, folate, biotin (all of which support brain metabolism in coordinated ways)
- Magnesium 200-400 mg/day — required cofactor for thiamine phosphorylation to TPP
- Address obvious risk factors — alcohol cessation if relevant, optimize glycemic control if diabetic, address sleep apnea if present, regular aerobic exercise
Tier 2 (MCI or early AD)
- Benfotiamine 300 mg twice daily (Gibson trial dose)
- Alpha lipoic acid 600 mg/day (R-ALA 300 mg/day if preferred)
- Acetyl-L-carnitine 1-2 g/day in divided doses
- Omega-3 fatty acids (DHA/EPA) 2-3 g/day — supports neuronal membrane function
- Vitamin D 2000-4000 IU/day if levels are low
- B12 (methylcobalamin) 1000-2000 mcg/day if levels are low or borderline
- Optimize HbA1c — the diabetic complications cascade contributes to cognitive decline
Tier 3 (severe baseline disease or strong family history)
- All of Tier 2, plus:
- Allithiamine (TTFD) 100-300 mg/day — the fat-soluble thiamine derivative with the best brain penetration; sometimes used in place of or in addition to benfotiamine for cognitive indications
- Consider thiamine status testing (erythrocyte transketolase activation coefficient, whole-blood TPP by HPLC) to confirm adequate repletion
- Coordinate with a neurologist or geriatrician familiar with the literature; some patients respond well to the protocol, others not
The protocol is conservative and supportive rather than aggressive. The evidence base does not yet justify claiming this combination prevents or treats AD, but it does represent a reasonable, low-risk approach to optimizing the metabolic substrate for brain function in patients with cognitive concerns.
Cautions
- This is not a substitute for standard AD evaluation and care — patients with new cognitive symptoms should have a full neurologic evaluation, including imaging, vascular risk assessment, B12/folate/thyroid screening, and consideration of conventional medications where indicated.
- The disease-modifying claim is not yet proven — the Gibson trial showed promising signals but is not definitive. Larger trials are underway. Until those report, the protocol should be framed as supportive rather than therapeutic.
- Mild cognitive impairment has many causes — depression, sleep apnea, medication side effects (anticholinergics, benzodiazepines), thyroid disease, vitamin deficiencies (B12, folate, D), normal pressure hydrocephalus, and others should all be evaluated before assuming AD is the cause.
- Severe alcohol use requires Wernicke prophylaxis first — any patient with active heavy alcohol use needs evaluation for occult thiamine deficiency and may need parenteral thiamine before any other intervention.
- B6 toxicity from combination products — some products combine benfotiamine with B6 at doses that, taken chronically, can cause B6-induced sensory neuropathy (paradoxically worsening symptoms). Keep B6 below 100 mg/day in chronic use.
- Drug interactions are minimal — benfotiamine and ALA do not significantly interact with most cognitive medications (donepezil, memantine, rivastigmine). They are generally safe to combine.
- Watch for hypoglycemia if on diabetes medications — ALA can lower blood glucose; benfotiamine is glucose-neutral. Monitor closely if adding ALA to insulin or sulfonylureas.
- Pair with magnesium — thiamine phosphorylation to TPP requires magnesium. Apparent failure of the protocol may reflect coexisting magnesium deficiency, which is common in older patients.
Key Research Papers
- Gibson GE et al. (2020). Benfotiamine and cognitive decline in Alzheimer's disease: results of a randomized placebo-controlled phase IIA clinical trial. J Alzheimers Dis. — PubMed
- Gibson GE, Blass JP (2007). Thiamine-dependent processes and treatment strategies in neurodegeneration. Antioxid Redox Signal. — PubMed
- Gibson GE et al. (1988). Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer's disease. Arch Neurol. — PubMed
- Mosconi L et al. (2008). FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer's disease. Eur J Nucl Med Mol Imaging. — PubMed
- Pan X et al. (2010). Powerful beneficial effects of benfotiamine on cognitive impairment and beta-amyloid deposition in amyloid precursor protein/presenilin-1 transgenic mice. Brain. — PubMed
- Pan X et al. (2016). Long-term cognitive improvement after benfotiamine administration in patients with Alzheimer's disease. Neurosci Bull. — PubMed
- Lu'o'ng KV, Nguyen LT (2011). Role of thiamine in Alzheimer's disease. Am J Alzheimers Dis Other Demen. — PubMed
- Bubber P et al. (2005). Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. — PubMed
- Mastrogiacoma F et al. (1996). Brain thiamine, its phosphate esters, and its metabolizing enzymes in Alzheimer's disease. Ann Neurol. — PubMed
- Ridley NJ et al. (2013). Alcohol-related dementia: an update of the evidence. Alzheimers Res Ther. — PubMed
- Sechi G et al. (2016). Advances in clinical determinants and neurological manifestations of B vitamin deficiency in adults. Nutr Rev. — PubMed
- Karuppagounder SS et al. (2009). Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer's mouse model. Neurobiol Aging. — PubMed
PubMed Topic Searches
- PubMed: thiamine + Alzheimer disease
- PubMed: benfotiamine + cognitive function
- PubMed: α-KGDH + Alzheimer
- PubMed: FDG-PET + AD hypometabolism
- PubMed: alcohol-related cognitive impairment + thiamine
Connections
- Vitamin B1 Overview
- B1 Benefits Hub
- B1 for Wernicke-Korsakoff
- B1 for Beriberi & Cardiac
- Benfotiamine for Diabetic Neuropathy
- Thiamine and Brain Health
- Alpha Lipoic Acid
- ALA for Mitochondria
- ALA for Neuroprotection
- Alzheimer's Disease
- Brain Fog
- Fatigue
- Vitamin B12
- Vitamin B6
- Magnesium
- Detox Protocols
- All Vitamins