Iron and Cognitive Performance
Iron is the rate-limiting cofactor for the brain's monoamine synthesis machinery — tyrosine hydroxylase (dopamine, norepinephrine) and tryptophan hydroxylase (serotonin) both require ferrous iron at their active sites — and it is required for the cholesterol-rich myelin sheath that oligodendrocytes wrap around axons to enable rapid saltatory conduction. The behavioral, attentional, and executive-function consequences of iron deficiency are therefore not secondary to anemia: they appear at the iron-deficient-erythropoiesis stage, before hemoglobin drops, and may not fully reverse in the developing brain even after later repletion. Betsy Lozoff's 25-year follow-up of Costa Rican infants with iron deficiency showed persistent IQ and behavioral deficits into early adulthood, the Murray-Kolb trials documented attention and verbal-memory gains in iron-deficient adult women restored to ferritin >30 ng/mL, and Earley and Allen demonstrated brain-region-specific iron loss as the central abnormality in restless legs syndrome. This deep-dive walks through the molecular machinery, the developmental windows, the adult ADHD/RLS literature, and the practical clinical thresholds.
Table of Contents
- Brain Iron Distribution and Why Some Regions Need More
- Tyrosine Hydroxylase, Dopamine, and Reward Circuitry
- Tryptophan Hydroxylase and Serotonin
- Myelin Synthesis and White-Matter Integrity
- The Lozoff Studies — Persistent Sequelae of Infant Iron Deficiency
- Murray-Kolb — Executive Function in Iron-Deficient Adult Women
- Ferritin and ADHD — The Konofal Signal
- Restless Legs Syndrome and the Allen Brain Ferritin Model
- Iron Deficiency in Cognitive Aging and Dementia Risk
- Clinical Thresholds and Practical Protocol
- Cautions (Brain Iron Excess in Neurodegeneration)
- Key Research Papers
- Connections
- Featured Videos
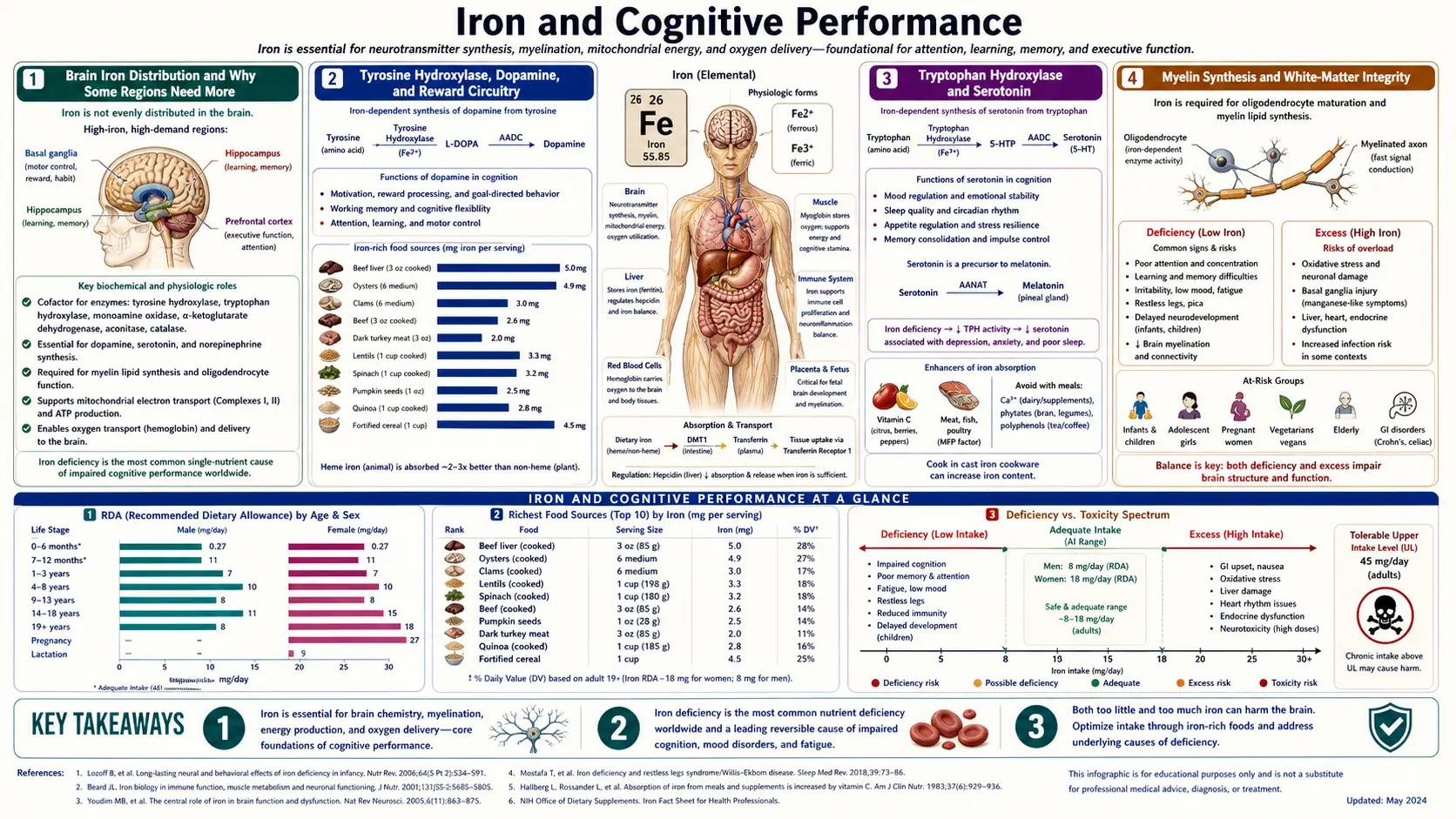
Brain Iron Distribution and Why Some Regions Need More
The adult human brain contains approximately 60 mg of non-heme iron, but it is not distributed evenly. The basal ganglia — specifically the substantia nigra, putamen, globus pallidus, and caudate nucleus — concentrate iron at 5 to 10 times the levels found in cortex. The choroid plexus, the red nucleus, and the dentate nucleus of the cerebellum are similarly iron-dense. These regions all share two features: high oxidative metabolism (making them energy-expensive and dependent on cytochrome-based ATP synthesis) and reliance on monoamine neurotransmitter signaling (making them dependent on the iron-requiring rate-limiting enzymes for dopamine and serotonin synthesis).
Iron crosses the blood-brain barrier through transferrin-receptor-mediated endocytosis at the endothelial cells of cerebral capillaries. Once inside the brain parenchyma, iron is shuttled by astrocyte-secreted transferrin to neurons and to oligodendrocytes, the latter being the most iron-rich cell type in the central nervous system. Oligodendrocytes need the iron not for their own dopaminergic signaling but to build the cholesterol- and lipid-rich myelin membranes they wrap around axons — iron is a cofactor for several enzymes in cholesterol biosynthesis and for the sphingolipid synthesis required for myelin assembly.
The brain regulates its own iron homeostasis somewhat independently of the systemic compartment. Hepcidin is expressed locally in the brain in response to inflammation and iron loading, and brain ferritin (predominantly the H-isoform with higher ferroxidase activity, suited to neurons' reactive-oxygen-prone environment) provides local storage. This means that systemic iron deficiency does not produce immediate proportional brain iron depletion — the brain is partially buffered — but sustained deficiency or deficiency during critical developmental windows will eventually deplete brain stores, particularly in the basal ganglia where demand is highest.
Tyrosine Hydroxylase, Dopamine, and Reward Circuitry
The rate-limiting step in dopamine synthesis is the hydroxylation of tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA), catalyzed by tyrosine hydroxylase (TH). TH belongs to the family of biopterin-dependent aromatic amino acid hydroxylases, and at its active site it carries a single ferrous (Fe2+) iron atom coordinated by two histidines and a glutamate. The iron atom activates molecular oxygen in cooperation with the tetrahydrobiopterin cofactor to produce a high-valent iron-oxo species that hydroxylates tyrosine. Without iron, the enzyme is catalytically dead. With insufficient iron, the enzyme works but does not keep up with dopaminergic firing demand — particularly in the nigrostriatal and mesocorticolimbic pathways that drive reward, motivation, motor initiation, and executive function.
The downstream metabolism is iron-dependent at multiple additional steps. Dopamine beta-hydroxylase, which converts dopamine to norepinephrine in noradrenergic neurons (locus coeruleus, ventral tegmental area, peripheral sympathetic terminals), is a copper enzyme but requires ascorbate-dependent iron reduction for cofactor recycling. Monoamine oxidase A and B, which degrade dopamine and norepinephrine in the synaptic cleft and intracellularly, are iron-containing flavoenzymes where the iron sits within the FAD environment.
Behaviorally, iron deficiency therefore produces a phenotype that overlaps strikingly with dopaminergic dysfunction: reduced motivation and reward sensitivity, attention deficits, impaired working memory, fatigue with intact muscle strength (suggesting a central rather than peripheral fatigue), and in extreme cases anhedonia. These are not vague subjective complaints — they map onto specific dopaminergic neural circuits and onto measurable behavioral testing on rotarod, eight-arm maze, and operant conditioning paradigms in iron-deficient animal models, all of which improve with iron repletion.
Tryptophan Hydroxylase and Serotonin
The rate-limiting enzyme in serotonin synthesis, tryptophan hydroxylase (TPH1 in the periphery, TPH2 in the brain), is structurally homologous to tyrosine hydroxylase and shares the same ferrous-iron / tetrahydrobiopterin active-site chemistry. It converts tryptophan to 5-hydroxytryptophan, which is then decarboxylated to serotonin by aromatic L-amino acid decarboxylase. Iron deficiency reduces TPH2 activity in the raphe nuclei, the midbrain origin of nearly all serotonergic projections to the cortex, hippocampus, and limbic system.
The serotonergic consequences of iron deficiency are less dramatic clinically than the dopaminergic ones — presumably because circulating tryptophan levels and TPH2 affinity for tryptophan provide partial buffering — but they contribute to the mood, sleep, and appetite changes documented in iron-deficient adults. Sleep architecture studies in iron-deficient infants show reduced REM sleep proportions, an effect partly attributable to disrupted serotonergic regulation of sleep-stage transitions. Mood symptoms (irritability, low mood, anxiety) in iron-deficient adolescent girls correlate with serum ferritin and improve with iron repletion in randomized trials.
The interaction with selective serotonin reuptake inhibitor therapy deserves more attention than it gets clinically. SSRIs work by blocking the serotonin transporter, increasing synaptic serotonin from whatever is being synthesized. If the rate-limiting synthesis step (TPH2) is iron-limited, an SSRI can only do so much. There is a small but consistent literature suggesting that iron repletion in iron-deficient patients with treatment-resistant depression improves SSRI response, and an open-label literature suggesting that iron status should be checked in any new depression workup before declaring a patient SSRI-non-responsive.
Myelin Synthesis and White-Matter Integrity
Oligodendrocytes are the most iron-rich cell type in the central nervous system, concentrating iron at levels several-fold higher than neurons. Their iron need is driven by their role as the brain's myelin factory. Each oligodendrocyte wraps multiple axons (up to 60 in human white matter) with successive lipid-rich membrane sheaths, and these membranes are 70-80% lipid by dry weight — primarily cholesterol, galactosylceramide, sulfatide, and phospholipids. Iron is a cofactor for several enzymes in cholesterol biosynthesis (including squalene monooxygenase and lanosterol 14-alpha-demethylase, both of which are iron-dependent oxygenases) and for the desaturases that introduce double bonds into myelin fatty acids.
Iron deficiency during the first two years of life — the period of most rapid myelination — produces measurable reductions in white-matter volume on MRI, reduced myelin water fraction, and slowed brainstem auditory evoked response conduction velocity. These structural abnormalities persist on follow-up MRI into adolescence even when serum ferritin has been normalized in the interim. This is the structural correlate of the persistent cognitive deficits observed in the Lozoff cohort discussed below.
The adult brain undergoes much less de novo myelination, but myelin turnover continues throughout life, and remyelination is a critical process after demyelinating injury (multiple sclerosis, stroke, traumatic brain injury). Iron deficiency in adults with multiple sclerosis is associated with poorer remyelination capacity and faster disability progression in observational cohorts, though the causal relationship has not been definitively established by intervention trial.
The Lozoff Studies — Persistent Sequelae of Infant Iron Deficiency
Betsy Lozoff's longitudinal cohort study of iron-deficient Costa Rican infants is the most-cited and most-influential body of work on the long-term cognitive consequences of early iron deficiency. The cohort was assembled in the 1980s and followed for over 25 years. At baseline, infants with iron deficiency anemia (hemoglobin < 10.5 g/dL with low serum ferritin and elevated FEP) were compared with iron-sufficient controls on the Bayley Scales of Infant Development. Anemic infants scored 10-12 points lower on the mental development index and 7-10 points lower on the psychomotor development index, with greater impairment in those whose deficiency was more severe and longer-standing at diagnosis.
The infants were then enrolled in a randomized trial of iron repletion. Critically, even after their hemoglobin and ferritin normalized in response to therapy and they were followed prospectively for years, the previously-anemic group continued to underperform on cognitive testing. At age 5, they scored lower on visual-motor integration, fine motor coordination, and standardized cognitive testing. At age 11-14, they scored lower on arithmetic, written expression, and motor function. At age 19, they were more likely to have not completed secondary school, to have repeated grades, and to report behavioral symptoms (anxiety, depression, social withdrawal) than the never-anemic controls.
The interpretation is that early iron deficiency during the critical period of brain development (peak myelination and synaptogenesis from approximately 6 months to 24 months postnatal) produces structural and functional changes that are not fully reversible by later iron repletion. The Costa Rica findings have been replicated in Chilean, Honduran, Indian, and Chinese cohorts. The mechanistic best-guess is reduced white-matter myelination at a non-recoverable developmental window, combined with persistent dopaminergic abnormalities in the developing basal ganglia.
The clinical implication is unambiguous: prevent iron deficiency during the first two years of life. The American Academy of Pediatrics recommends universal screening for iron deficiency at age 12 months, iron-fortified formula or iron supplementation for breastfed infants from 4 months until complementary foods provide adequate iron, and iron-fortified cereals as an early complementary food. Routine ferrous sulfate drops (1 mg/kg/day from 4 months in exclusively breastfed infants) are inexpensive and effective.
Murray-Kolb — Executive Function in Iron-Deficient Adult Women
Laura Murray-Kolb and colleagues at Penn State conducted a series of randomized, placebo-controlled iron-supplementation trials in iron-deficient (but not necessarily anemic) adult women to test whether the cognitive deficits associated with iron deficiency could be reversed with repletion in adults — a question previously assumed to be answered yes but never rigorously tested in adult cohorts.
In the seminal 2007 trial, 113 non-anemic iron-deficient women (ages 18-35, ferritin < 16 ng/mL, hemoglobin normal) were randomized to ferrous sulfate (160 mg iron daily) or placebo for 16 weeks. At baseline, the iron-deficient women performed worse on attention, memory, and learning tasks than a separately recruited iron-sufficient comparison group. After supplementation, the previously iron-deficient women who achieved repletion (ferritin to > 30 ng/mL) showed significant improvements on cognitive testing — the magnitude of improvement scaling with the magnitude of ferritin rise. The placebo group showed no comparable improvements.
Specifically, the cognitive domains showing the largest improvements were: attention (5-task continuous performance), verbal learning (Rey Auditory Verbal Learning Test), memory (delayed recall), and processing speed. Executive function on tests of working memory and task-switching also improved. The effect sizes were clinically meaningful — comparable to the effect size of methylphenidate in mild adult ADHD.
The key clinical takeaway is that the cognitive symptoms attributed to "just being tired" or "stressed" in young adult women, particularly those with heavy menstrual periods, frequent blood donation, or vegetarian/vegan diets, may be substantially due to iron-deficient cognition even in the absence of anemia. The diagnostic threshold should be ferritin, not hemoglobin. A premenopausal woman with ferritin in the 10-20 ng/mL range and persistent fatigue, brain fog, or attention complaints is a candidate for therapeutic iron repletion regardless of whether her hemoglobin meets WHO criteria for anemia.
Ferritin and ADHD — The Konofal Signal
Eric Konofal and colleagues at Hôpital Robert-Debré in Paris reported in 2004 that children with attention-deficit/hyperactivity disorder had significantly lower serum ferritin levels than age-matched controls (mean 23 ng/mL vs 44 ng/mL), and that ferritin level inversely correlated with ADHD symptom severity on the Conners Parent Rating Scale. The relationship was independent of hemoglobin — the ADHD children were not anemic, only iron-low — suggesting that brain iron status (which ferritin partially indexes) was the relevant variable, not erythrocyte iron.
The follow-up open-label trial by the same group tested ferrous sulfate (80 mg/day) in 23 ADHD children with ferritin < 30 ng/mL for 12 weeks. ADHD symptom scores on Conners and the ADHD Rating Scale improved progressively over the trial period, with effect sizes comparable to a low-dose stimulant trial. Subsequent placebo-controlled trials by other groups have produced mixed results, with some confirming the Konofal signal and others (notably in non-iron-deficient ADHD populations) finding no benefit.
The consensus interpretation is that ADHD in the context of low brain iron is iron-responsive, but ADHD per se is not an iron-deficiency disease. The clinical implication: any child or adult being evaluated for ADHD should have ferritin checked. If ferritin is below 30 ng/mL, iron repletion is the appropriate first-line intervention before or alongside stimulant trial. If ferritin is adequate, iron supplementation will not improve ADHD symptoms and should not be expected to. The same principle applies to adult ADHD — ferritin screening is a low-cost, high-information addition to the standard workup.
Mechanistically, the ADHD-iron connection runs through the dopaminergic pathways discussed above. ADHD is increasingly understood as a dopamine-deficit-and-dysregulation syndrome in the prefrontal-striatal circuits, and stimulant medications work by elevating synaptic dopamine. If tyrosine hydroxylase is iron-limited, the dopamine deficit is partly a biosynthetic one, and iron repletion addresses the upstream supply problem rather than just the downstream reuptake problem.
Restless Legs Syndrome and the Allen Brain Ferritin Model
Restless legs syndrome (RLS), now also called Willis-Ekbom disease, is one of the clearest examples of region-specific brain iron deficiency producing a clinically distinct neurological syndrome. Richard Allen and colleagues at Johns Hopkins published the foundational evidence in the early 2000s: cerebrospinal fluid ferritin is reduced and CSF transferrin elevated in RLS patients compared with controls, despite normal serum iron studies. Subsequent post-mortem and MRI studies confirmed reduced iron content specifically in the substantia nigra of RLS patients.
The pathophysiology is now understood as a regional dopaminergic deficit in nigrostriatal and diencephalic A11 dopamine pathways, driven by reduced tyrosine hydroxylase activity from regional iron deficiency, with downstream consequences for descending modulation of spinal sensorimotor processing. The clinical syndrome — an irresistible urge to move the legs that worsens at rest, in the evening, and in bed, and is partially relieved by movement — reflects this circuit-level dysfunction. RLS is dramatically responsive to dopaminergic medications (pramipexole, ropinirole) for the same reason.
The clinical implication is that every patient with RLS should have ferritin checked, and ferritin should be optimized to > 75 ng/mL (some experts say > 100) before declaring the patient a dopaminergic-agonist candidate. The reasoning: iron repletion can dramatically reduce or eliminate RLS symptoms in iron-deficient patients without exposing them to the long-term complications of dopaminergic medications (augmentation, impulse control disorders). The Allen group has shown that intravenous ferric carboxymaltose can produce sustained RLS improvement when oral iron is insufficient or poorly tolerated — an approach now incorporated into the International Restless Legs Syndrome Study Group consensus guidelines for iron-deficient RLS.
RLS in pregnancy, a common and often overlooked entity, is particularly amenable to iron-based management because the pregnancy itself produces iron depletion that worsens through the third trimester. Aggressive iron supplementation in pregnant women with RLS often resolves the syndrome without need for medication.
Iron Deficiency in Cognitive Aging and Dementia Risk
The cognitive role of iron in older adults is more complicated than in young adults or infants, because the aging brain accumulates iron in pathological deposits while sometimes being functionally iron-deficient at the cellular level. Excess iron in the substantia nigra is a feature of Parkinson's disease, and excess iron in the basal ganglia, hippocampus, and cortex has been documented in Alzheimer's disease postmortem and on susceptibility-weighted MRI.
At the same time, frank iron-deficiency anemia in older adults is associated with accelerated cognitive decline, increased dementia risk in observational cohorts, and worse outcomes after stroke and hip fracture. The reconciliation is that iron in the wrong place (deposited in plaques, in degenerating neurons, in inflammatory microglia) is toxic, while iron in the right place (in healthy neurons supporting normal neurotransmitter synthesis) is essential. Systemic iron-deficiency anemia in the elderly does not improve neurodegeneration but does worsen the global cognitive picture by producing brain hypoxia, reduced ATP supply, and impaired neurotransmitter synthesis on top of any underlying neurodegenerative process.
The practical implication for older adults: iron-deficiency anemia should be treated like any other treatable contributor to cognitive impairment, but iron supplementation should not be given to non-deficient older adults in the hope of improving brain function, and patients with documented neurodegenerative disease (PD, AD) should have ferritin and transferrin saturation kept in the lower-normal range (50-100 ng/mL, <40% sat) rather than pushed toward repletion targets used in younger adults.
Clinical Thresholds and Practical Protocol
For brain-focused iron repletion, the relevant biomarker is serum ferritin, not hemoglobin. Hemoglobin is a lagging indicator that does not drop until the deficiency is severe. Ferritin is the body's storage iron and the best proxy for brain iron availability.
- Ferritin < 30 ng/mL: Iron-deficient by any clinical definition. Treat. Pregnant women, RLS patients, ADHD patients, and patients with cognitive complaints should target ferritin > 50 (general adult) or > 75-100 (RLS, treatment-resistant depression, severe ADHD).
- Ferritin 30-50 ng/mL: "Adequate" by population norms but probably suboptimal for brain-iron-dependent functions in symptomatic patients. Consider repletion trial in patients with brain-symptom complaints (cognitive fog, attention deficits, mood, restless legs, fatigue).
- Ferritin 50-150 ng/mL: Optimal for most adults. No supplementation needed.
- Ferritin > 200 ng/mL (women) or > 300 ng/mL (men): Possible iron overload. Investigate (HFE gene testing, transferrin saturation, liver enzymes). Do not supplement.
The first-line iron supplement remains ferrous sulfate 325 mg (65 mg elemental iron) on alternate days (per the alternate-day-dosing evidence discussed on the Iron Deficiency Anemia page) taken with vitamin C 250 mg, away from coffee/tea/calcium/dairy. Ferrous bisglycinate is a well-tolerated alternative for patients with GI intolerance to sulfate. Recheck ferritin at 8-12 weeks; expect a rise of 25-50 ng/mL per quarter on therapy if the underlying deficiency is corrected.
Cautions (Brain Iron Excess in Neurodegeneration)
- Neurodegenerative iron excess: Pantothenate kinase-associated neurodegeneration (PKAN, formerly Hallervorden-Spatz disease), neuroferritinopathy, aceruloplasminemia, and the "neurodegeneration with brain iron accumulation" (NBIA) spectrum all involve pathological iron deposition in basal ganglia. Iron supplementation is contraindicated. So is empirical iron in any patient with established Parkinson's or Alzheimer's without documented systemic iron deficiency.
- Morley Robbins / copper-ceruloplasmin framework: The Robbins critique of iron supplementation argues that much of what is called "iron deficiency" is actually copper deficiency producing functional iron deficiency, because copper-dependent ceruloplasmin is required to load iron onto transferrin and to mobilize iron from ferritin stores. In this view, giving iron without addressing copper insufficiency drives unbound iron deposition into tissues, including brain, while leaving the underlying ceruloplasmin functional deficit untreated. See Hemoglobin and Ceruloplasmin and Whole Food Copper Sources for this counterpoint. Clinical practice: check ceruloplasmin alongside ferritin and transferrin saturation if iron supplementation does not produce expected ferritin rise.
- Hemochromatosis screening: Anyone of Northern European ancestry being started on chronic iron supplementation should have HFE genotyping (C282Y, H63D) and baseline transferrin saturation. A heterozygote C282Y carrier supplemented with iron can accelerate iron overload.
- SSRI / dopaminergic interactions: Iron repletion in iron-deficient patients on stimulants or dopaminergic agonists can occasionally unmask dopamine excess (anxiety, agitation, insomnia). Start at lower doses and titrate. The same caution applies to iron repletion in patients on SSRIs — some develop transient activation or akathisia as serotonin synthesis catches up with reuptake blockade.
- Constipation and GI tolerability: The leading cause of iron-supplementation failure is GI intolerance. Alternate-day dosing, taking iron with food (at the cost of some absorption), ferrous bisglycinate, or splitting to smaller divided doses all help.
Key Research Papers
- Lozoff B, Jimenez E, Hagen J, Mollen E, Wolf AW (2000). Poorer behavioral and developmental outcome more than 10 years after treatment for iron deficiency in infancy. Pediatrics. — PubMed
- Lozoff B, Beard J, Connor J, Felt B, Georgieff M, Schallert T (2006). Long-lasting neural and behavioral effects of iron deficiency in infancy. Nutrition Reviews. — PubMed
- Murray-Kolb LE, Beard JL (2007). Iron treatment normalizes cognitive functioning in young women. American Journal of Clinical Nutrition. — PubMed
- Konofal E, Lecendreux M, Arnulf I, Mouren MC (2004). Iron deficiency in children with attention-deficit/hyperactivity disorder. Archives of Pediatrics and Adolescent Medicine. — PubMed
- Konofal E, Lecendreux M, Deron J, Marchand M, Cortese S et al. (2008). Effects of iron supplementation on attention deficit hyperactivity disorder in children. Pediatric Neurology. — PubMed
- Allen RP, Earley CJ (2007). The role of iron in restless legs syndrome. Movement Disorders. — PubMed
- Earley CJ, Connor J, Garcia-Borreguero D, Jenner P, Winkelman J et al. (2014). Altered brain iron homeostasis and dopaminergic function in Restless Legs Syndrome. Sleep Medicine. — PubMed
- Allen RP, Picchietti DL, Auerbach M et al. (2018). Evidence-based and consensus clinical practice guidelines for the iron treatment of restless legs syndrome (IRLSSG, EURLSSG, RLS-Foundation). Sleep Medicine. — PubMed
- Beard JL, Connor JR (2003). Iron status and neural functioning. Annual Review of Nutrition. — PubMed
- Georgieff MK (2011). Long-term brain and behavioral consequences of early iron deficiency. Nutrition Reviews. — PubMed
- Wenger MJ, DellaValle DM, Murray-Kolb LE, Haas JD (2019). Effect of iron deficiency on simultaneous measures of behavior, brain activity, and energy expenditure in the performance of a cognitive task. Nutritional Neuroscience. — PubMed
- Beard J (2003). Iron deficiency alters brain development and functioning. Journal of Nutrition. — PubMed
- Murray-Kolb LE (2011). Iron status and neuropsychological consequences in women of reproductive age: what do we know and where are we headed? Journal of Nutrition. — PubMed
PubMed Topic Searches
- PubMed: Iron deficiency adult cognition
- PubMed: Ferritin and ADHD
- PubMed: RLS iron ferritin
- PubMed: TH iron dopamine
- PubMed: Infant iron myelination
Connections
- Iron Benefits Hub
- Iron Overview
- Iron Deficiency Anemia
- Heme vs Non-Heme Iron
- Iron and Athletic Performance
- Copper
- Hemoglobin and Ceruloplasmin
- Zinc
- Vitamin C
- Vitamin B12
- Folate (B9)
- L-Tyrosine
- L-Tryptophan
- Fatigue
- Restless Legs Syndrome
- Parkinson's Disease
- Iron Panel
- Whole Food Copper Sources
- Organ Meats