Copper for Hemoglobin and Ceruloplasmin
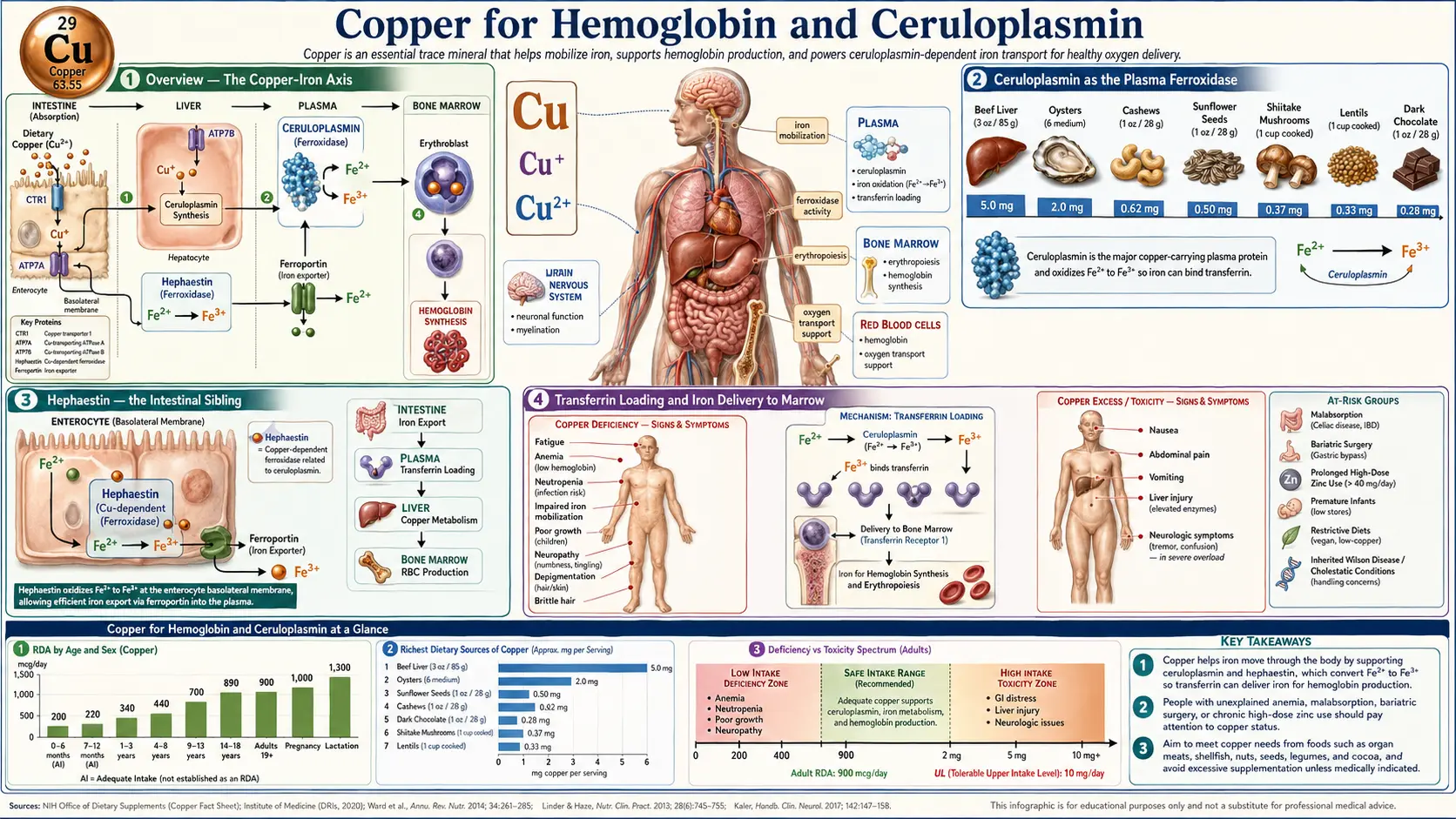
The single most clinically important fact about copper is that iron without copper does not become hemoglobin. The copper-dependent enzyme ceruloplasmin (and its membrane-bound intestinal partner hephaestin) oxidizes Fe²+ to Fe³+, the only iron oxidation state that can bind transferrin and reach the bone marrow. A patient with adequate iron stores but inadequate ceruloplasmin activity will present with a classic iron-deficiency-pattern microcytic anemia that does not respond to iron supplementation, alongside paradoxical tissue iron accumulation in liver and macrophages. This page walks through the ceruloplasmin–transferrin–hemoglobin axis, the genetic disease (aceruloplasminemia) that proves the requirement, the acquired causes of functional copper deficiency (zinc overdose, denture cream, bariatric surgery), and the Morley Robbins / Root Cause Protocol framework that has popularized this biochemistry in the alternative-medicine literature.
Table of Contents
- Overview — The Copper-Iron Axis
- Ceruloplasmin as the Plasma Ferroxidase
- Hephaestin — the Intestinal Sibling
- Transferrin Loading and Iron Delivery to Marrow
- Hemoglobin Synthesis and the Heme Pathway
- Iron Without Copper — the Functional Iron-Deficiency Paradox
- Aceruloplasminemia — the Genetic Proof
- Acquired Causes of Functional Copper Deficiency
- The Morley Robbins / Root Cause Protocol Framework
- Testing — Serum Copper vs Ceruloplasmin vs Bioavailable Copper
- Treatment — Repleting Functional Copper
- Key Research Papers
- Connections
- Featured Videos
Overview — The Copper-Iron Axis
The textbook picture of hematology — iron in, hemoglobin out — obscures the fact that iron itself cannot move from the gut into circulation, or from macrophages back into circulation after a red blood cell is recycled, without a copper-dependent enzyme doing the chemistry that allows transferrin to pick it up. The copper requirement was first systematically demonstrated by George Cartwright and Maxwell Wintrobe at the University of Utah in the late 1940s and 1950s, working with copper-deficient pigs that developed a profound anemia that did not respond to iron supplementation but resolved within days of copper repletion. Their work, and parallel studies in copper-deficient infants on cow-milk formula (Cordano A et al. 1964), established the modern understanding that iron metabolism is fundamentally copper-dependent.
The mechanism is now well-mapped: copper is the catalytic redox center in two distinct ferroxidase enzymes — the soluble plasma protein ceruloplasmin (which contains six copper atoms per molecule) and the membrane-bound intestinal protein hephaestin. Both perform the same chemistry: oxidation of ferrous Fe²+ to ferric Fe³+. This is mandatory because the iron transport protein transferrin binds only Fe³+, not Fe²+. Without ferroxidase activity, iron exits the enterocyte through ferroportin in the Fe²+ state and either oxidizes spontaneously and slowly to a form transferrin can bind, or (more commonly) it backs up in the enterocyte and gets shed back into the lumen when the cell is sloughed at the end of its 3-5 day lifespan.
Ceruloplasmin as the Plasma Ferroxidase
Ceruloplasmin is a 132-kilodalton plasma glycoprotein synthesized in the liver, containing six copper atoms per molecule arranged in three type I, one type II, and two type III copper centers. Approximately 90–95% of all circulating copper in plasma is bound to ceruloplasmin; the remainder is loosely bound to albumin, transcuprein (alpha-2-macroglobulin), and amino acids (the "free" or "bioavailable" copper pool measured by the difference between serum copper and ceruloplasmin-bound copper).
The ferroxidase activity is the most studied of ceruloplasmin's functions. The reaction is:
4 Fe²+ + O&sub2; + 4 H+ → 4 Fe³+ + 2 H&sub2;O
Each ceruloplasmin molecule catalyzes this reaction at roughly 600–800 iron atoms per minute under physiologic conditions. The Fe³+ product is the substrate transferrin requires — transferrin's two iron-binding sites have an affinity for Fe³+ that is roughly 10²0; times greater than for Fe²+, so the oxidation step is what gates iron mobilization from macrophages (recycling senescent red cells), from hepatocytes (releasing iron stores from ferritin), and across the basolateral membrane of enterocytes.
Ceruloplasmin also has minor copper-transport activity, oxidase activity against biogenic amines, and antioxidant activity against superoxide and hydrogen peroxide — functions that overlap with the antioxidant role of copper described on the antioxidant defense page. But the ferroxidase activity is the rate-limiting one for hematology.
Hephaestin — the Intestinal Sibling
Hephaestin (named for the Greek god of metalworking) is a membrane-bound copper-containing ferroxidase homologous to ceruloplasmin, expressed on the basolateral membrane of intestinal enterocytes. Where ceruloplasmin performs ferroxidase chemistry in plasma, hephaestin performs the same chemistry at the moment iron exits the enterocyte through the ferroportin channel into the portal circulation.
The hephaestin–ferroportin pairing was definitively demonstrated by the sex-linked anemia (sla) mouse, a naturally occurring hephaestin loss-of-function mutation that causes iron-deficiency anemia despite normal intestinal iron uptake. The sla mouse absorbs iron normally across the apical brush border (via DMT1) and the iron accumulates inside the enterocyte, but cannot be efficiently exported across the basolateral membrane because there is no ferroxidase to oxidize it. The mouse then loses the iron when the enterocyte is shed.
In humans, hephaestin mutations are rare, but acquired hephaestin deficiency from severe copper depletion produces the same picture. This is why copper-deficient patients on parenteral nutrition or after bariatric surgery sometimes have normal serum iron studies (because iron is sitting inside enterocytes and macrophages) but profound microcytic anemia.
Transferrin Loading and Iron Delivery to Marrow
Once Fe³+ is generated by ceruloplasmin or hephaestin, it binds transferrin in the plasma. Each transferrin molecule has two high-affinity Fe³+-binding sites and circulates with a typical saturation of 25–35%. The iron-saturated holotransferrin is then taken up by erythroid precursors in the bone marrow via the transferrin receptor (TfR1, also called CD71), which is highly expressed on developing red cells exactly because of their enormous iron demand for hemoglobin synthesis.
Inside the erythroid precursor, transferrin and iron are internalized by clathrin-mediated endocytosis. The endosome acidifies, releases iron from transferrin, and the iron is transported through DMT1 in the endosomal membrane into the cytosol. Apotransferrin is then recycled back to the cell surface and released to find more iron. The cytosolic iron is delivered to the mitochondria, where it enters the protoporphyrin IX ring to form heme, the prosthetic group of hemoglobin.
The entire pipeline — macrophage iron recycling, intestinal iron absorption, transferrin loading, erythroid receptor uptake, mitochondrial heme synthesis — depends on the upstream ferroxidase step performed by copper-dependent enzymes. Lose that step and the whole pipeline backs up.
Hemoglobin Synthesis and the Heme Pathway
Hemoglobin is a tetramer of two alpha and two beta globin chains, each binding one heme prosthetic group at the center, for four total heme groups per hemoglobin molecule. The heme group is a protoporphyrin IX ring with a central iron atom coordinated to four pyrrole nitrogens, a proximal histidine residue from the globin chain, and (when oxygenated) a molecular oxygen molecule. The iron must be in the Fe²+ state for oxygen binding (Fe³+ heme is methemoglobin and does not bind oxygen).
The heme synthesis pathway has eight enzymatic steps. The first and rate-limiting step (delta-aminolevulinic acid synthase, ALAS) occurs in the mitochondrion, the next four in the cytosol, and the final three back in the mitochondrion. The final step — ferrochelatase — inserts Fe²+ into protoporphyrin IX to produce heme. Ferrochelatase requires the iron in the Fe²+ oxidation state, so even though ceruloplasmin's ferroxidase produces Fe³+ for transferrin transport, the iron is reduced back to Fe²+ (by the still-incompletely-characterized mitochondrial reductases) for heme insertion. The reduction is not the rate-limiting step; the upstream copper-dependent oxidation for transferrin loading is.
The clinical implication: when copper is the limiting factor, the hematologic picture mimics iron deficiency exactly — low hemoglobin, microcytic mean corpuscular volume (MCV), low mean corpuscular hemoglobin (MCH), reactive elevation of red cell distribution width (RDW), and reactive thrombocytosis. The clue that distinguishes copper-deficient from iron-deficient anemia is concomitant neutropenia (rare in pure iron deficiency, common in copper deficiency) and the failure of the anemia to respond to oral or even IV iron.
Iron Without Copper — the Functional Iron-Deficiency Paradox
The classic clinical scenario is a patient with persistent iron-deficiency-pattern anemia that does not respond to high-dose iron supplementation. The standard hematologic workup — ferritin, serum iron, transferrin saturation, total iron-binding capacity — often shows a confusing picture. Ferritin (the intracellular iron storage protein) may be elevated, because iron is accumulating inside hepatocytes and macrophages but cannot be mobilized for hemoglobin synthesis. Serum iron may be low, normal, or even elevated. Transferrin saturation is often paradoxically reduced because transferrin keeps circulating without sufficient iron loading.
The Morley Robbins framework formalizes this paradox: in many modern patients, the issue is not iron deficiency in the classical sense but iron dysregulation — tissue iron overload coexisting with functional iron deficiency at the bone marrow, all driven by inadequate ceruloplasmin activity. The proposed treatment is to address the copper, retinol, and magnesium status that gate ceruloplasmin synthesis, rather than to administer more iron (which Robbins argues makes the situation worse by adding to the tissue iron burden without addressing the mobilization defect).
The conventional medical literature does not fully endorse this framing — many anemic patients are genuinely iron-deficient and respond promptly to iron supplementation — but the copper-deficient subgroup is genuine and underdiagnosed. The key clinical clue is the combination of microcytic anemia plus unexplained neutropenia (sometimes with reactive thrombocytopenia) in a patient with risk factors: zinc overdose, denture cream use, bariatric surgery, gastric bypass, prolonged parenteral nutrition, or longstanding malabsorption (celiac, Crohn's).
Aceruloplasminemia — the Genetic Proof
Aceruloplasminemia is the autosomal recessive loss-of-function disorder of the ceruloplasmin gene, first characterized by Hidehiro Miyajima in Japan in the late 1980s. Patients have undetectable serum ceruloplasmin and ferroxidase activity. The clinical picture, which usually emerges in the third to fifth decade of life, demonstrates exactly what happens when ferroxidase activity is absent:
- Microcytic anemia — mild to moderate, typically refractory to iron
- Tissue iron overload — in liver (cirrhosis), pancreas (insulin-dependent diabetes mellitus from beta-cell destruction), brain (basal ganglia and dentate nucleus iron deposition visible on MRI as low T2 signal)
- Neurodegenerative disease — movement disorder (parkinsonism, dystonia, chorea), cerebellar ataxia, cognitive decline
- Retinal degeneration — from RPE iron toxicity
The aceruloplasminemia phenotype is the most extreme illustration of the copper-iron axis. Patients have plenty of iron — they have a pathologic excess of tissue iron — but cannot use it for hemoglobin synthesis because the ferroxidase step is absent. The neurodegenerative consequences arise because free Fe²+ in tissue catalyzes Fenton-chemistry hydroxyl-radical generation (see the antioxidant defense page), and the brain's lipid-rich, high-oxidative-demand environment is particularly vulnerable.
Aceruloplasminemia treatment is iron chelation (deferoxamine or deferiprone), not iron supplementation. Ceruloplasmin replacement is not yet clinically available. The disease serves as nature's demonstration of why copper is mandatory for safe iron handling.
Acquired Causes of Functional Copper Deficiency
Far more common than genetic aceruloplasminemia is acquired copper deficiency from any of several modern causes:
- Zinc overdose — zinc and copper compete for the intestinal copper transporter and for binding to metallothionein in the enterocyte. Sustained zinc intake above ~50 mg/day (especially the 100 mg/day "immune support" megadoses) saturates intestinal metallothionein with zinc, which then preferentially binds incoming copper and prevents its absorption. The Mayo Clinic series (Kumar N et al.) found zinc-induced copper deficiency myelopathy in patients taking zinc lozenges for chronic cold prevention, oral zinc for taste disorders, or zinc-containing denture creams.
- Denture cream (Fixodent, Poligrip) — older formulations contained zinc as a binding agent. Patients using multiple applications per day for years accumulated systemic zinc burden sufficient to deplete copper. After class-action litigation, the manufacturers reformulated to zinc-free versions in 2010, but legacy cases remain.
- Bariatric surgery (Roux-en-Y gastric bypass, duodenal switch) — bypass of the duodenum and proximal jejunum, where copper absorption is concentrated, produces copper deficiency in 10–20% of bariatric patients over 5–10 years. Routine surveillance and prophylactic copper supplementation is now standard post-bariatric care, but many patients fall out of follow-up and present years later with myelopathy and anemia.
- Prolonged parenteral nutrition — copper requirements may be underdosed in long-term TPN, especially in pediatric and neonatal patients. Cholestatic liver disease in TPN patients can complicate copper management because biliary copper excretion is impaired.
- Severe malabsorption (celiac disease, Crohn's, short-bowel syndrome) — reduces copper absorption proportional to the affected intestinal length.
- Anti-copper chelation therapy — D-penicillamine, trientine, or zinc therapy for Wilson disease (or for rheumatoid arthritis, in the historical penicillamine era) can produce iatrogenic copper deficiency if monitoring is inadequate.
The shared feature is that the patient looks fine for years and then presents with a slowly progressive myelopathy (gait ataxia, sensory loss, spasticity) plus refractory cytopenias. Recognition is the hard part — once copper is checked and replaced, hematologic recovery is usually rapid and complete, but neurologic recovery is often partial because axonal demyelination has already occurred.
The Morley Robbins / Root Cause Protocol Framework
Morley Robbins is a former pharmacist, the founder of the Magnesium Advocacy Group, and the architect of the Root Cause Protocol. Over the last decade his work has popularized, in the alternative-medicine and functional-medicine communities, the proposition that a substantial fraction of modern chronic disease — fatigue, anemia, neurodegeneration, autoimmunity, even some cancers — traces to functional copper deficiency masquerading as iron deficiency. The framework rests on the biochemistry described above: that ceruloplasmin activity gates iron mobilization, that magnesium and retinol-form Vitamin A are required for hepatic ceruloplasmin synthesis, and that the typical Western diet (low in beef liver and oysters, high in fortified iron from refined grains) produces iron loading without proportional copper loading.
The Robbins recommendations include:
- Stop synthetic iron supplementation unless absolutely necessary — in his framing, fortified iron drives tissue iron accumulation without resolving the bone-marrow mobilization defect.
- Whole-food copper from beef liver (1–3 oz per week), oysters (the highest copper concentration of any common food), cacao / dark chocolate, bee pollen, and (controversially) bovine adrenal cortex — see Whole Food Copper Sources.
- Magnesium repletion — Robbins argues that magnesium is essential for ATP-dependent copper loading onto apoceruloplasmin in the liver.
- Retinol-form Vitamin A (not beta-carotene) from beef liver or cod liver oil — required for hepatic ceruloplasmin synthesis via the RAR/RXR nuclear receptor pathway.
- Avoid high-dose zinc supplementation — the copper-depleting effect described above.
- Test ceruloplasmin enzyme activity, not just serum copper, to identify functional deficiency.
The conventional medical literature has not formally validated the Robbins framework as a unified theory of chronic disease, but the individual claims about ceruloplasmin biochemistry are mainstream. The pages on Morley Robbins, Ceruloplasmin and Bioavailable Copper, Copper-Iron Dysregulation, and Iron Overload Hidden Toxicity develop the framework in more detail.
Testing — Serum Copper vs Ceruloplasmin vs Bioavailable Copper
Three different laboratory measurements address copper status, and they answer different questions:
- Serum copper — total copper in plasma, including ceruloplasmin-bound (~90%) and loosely bound to albumin and amino acids (~10%). Normal range approximately 70–150 mcg/dL in adults. Elevated in acute phase reaction (ceruloplasmin is an acute-phase reactant), pregnancy, oral contraceptive use, and Wilson disease (sometimes). Low in copper deficiency.
- Ceruloplasmin (immunoassay or enzymatic activity) — the protein concentration (typically 20–60 mg/dL) or its enzyme activity. The immunoassay measures total ceruloplasmin protein; the enzymatic assay measures functional ferroxidase activity. The two can dissociate — some patients have apoceruloplasmin (the copper-free precursor protein) that registers normal on immunoassay but lacks ferroxidase activity. The enzymatic activity is more functionally meaningful but harder to find at most labs.
- Bioavailable (non-ceruloplasmin) copper — calculated as serum copper minus the copper bound to ceruloplasmin (each ceruloplasmin molecule binds 6 copper atoms; ceruloplasmin copper = 3 × ceruloplasmin in mg/dL, in mcg/dL). Normal <15% of total serum copper. Elevated in Wilson disease and other copper overload states. Low or low-normal in functional copper deficiency.
For diagnosis of acquired copper-deficiency myelopathy or copper-deficient anemia, the most useful battery is serum copper, ceruloplasmin (immunoassay), serum zinc, complete blood count with differential, ferritin, B12, methylmalonic acid, and homocysteine. The B12 testing is essential because the clinical picture overlaps so completely with B12 deficiency that the two are often misdiagnosed as each other.
Treatment — Repleting Functional Copper
Once copper deficiency is identified, treatment is straightforward:
- Identify and remove the cause — discontinue zinc supplements, switch to zinc-free denture cream, treat malabsorption, optimize parenteral nutrition.
- Oral copper repletion — copper sulfate, copper gluconate, or copper bisglycinate at 2–8 mg elemental copper per day (the RDA is 0.9 mg/day; repletion doses are 2–10× RDA for 6–12 weeks then taper to maintenance). Cu Bisglycinate is well-tolerated; copper sulfate can be GI-irritating.
- Whole-food sources — beef liver (one 3 oz serving provides ~10–15 mg copper, far above any oral supplement), oysters (the highest copper density of any common food at ~3–5 mg per 100 g for raw eastern oysters), cocoa / dark chocolate (~1 mg per ounce of 70% or higher), cashews, sunflower seeds, sesame seeds, mushrooms (shiitake especially), and spirulina. See Whole Food Copper Sources.
- Cofactor support — ensure adequate magnesium and retinol-form Vitamin A for hepatic ceruloplasmin synthesis.
- Monitor — serum copper, ceruloplasmin, and CBC at 6 weeks and 3 months. Hematologic recovery is usually rapid (reticulocytosis within days, hemoglobin normalization within weeks). Neurologic recovery is slower and often incomplete.
- Avoid IV copper outside of TPN settings — oral repletion is almost always adequate and safer.
For the related issue of iron management in copper-deficient patients (when to use iron supplementation, when to chelate), see our pages on Anemia, Iron, and Iron Deficiency Anemia.
Key Research Papers
- Cartwright GE, Wintrobe MM (1964). Copper metabolism in normal subjects. American Journal of Clinical Nutrition. — PubMed
- Cordano A, Baertl JM, Graham GG (1964). Copper deficiency in infancy. Pediatrics. — PubMed
- Harris ZL et al. (1995). Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. PNAS. — PubMed
- Miyajima H et al. (1987). Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration. Neurology. — PubMed
- Vulpe CD et al. (1999). Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nature Genetics. — PubMed
- Kumar N et al. (2003). Copper deficiency myelopathy (human swayback). Mayo Clinic Proceedings. — PubMed
- Nemeth E, Ganz T (2006). Regulation of iron metabolism by hepcidin. Annual Review of Nutrition. — PubMed
- Olivares M, Uauy R (1996). Copper as an essential nutrient. American Journal of Clinical Nutrition. — PubMed
- Reeves PG, DeMars LCS (2004). Copper deficiency reduces iron absorption and biological half-life in male rats. Journal of Nutrition. — PubMed
- Spinazzi M et al. (2007). Myelo-optico-neuropathy in copper deficiency occurring after partial gastrectomy. Journal of Neurology. — PubMed
- Halfdanarson TR et al. (2008). Hematological manifestations of copper deficiency: a retrospective review. European Journal of Haematology. — PubMed
- Nations SP et al. (2008). Denture cream: an unusual source of excess zinc, leading to hypocupremia and neurologic disease. Neurology. — PubMed
PubMed Topic Searches
- PubMed: Ceruloplasmin ferroxidase
- PubMed: Copper-deficiency anemia
- PubMed: Aceruloplasminemia
- PubMed: Hephaestin and iron export
- PubMed: Copper-iron axis
Connections
- Copper Overview
- Copper Benefits Hub
- Copper for Connective Tissue
- Copper for Antioxidant Defense
- Copper for Neurological Health
- Iron
- Iron Deficiency Anemia
- Heme vs Non-Heme Iron
- Zinc
- Magnesium
- Morley Robbins
- Ceruloplasmin and Bioavailable Copper
- Copper-Iron Dysregulation
- Whole Food Copper Sources
- Iron Overload Hidden Toxicity
- Vitamin A (Required for Ceruloplasmin Synthesis)
- Vitamin C
- Vitamin B12
- Anemia
- Beef
- Organ Meats (Beef Liver)
- Dark Chocolate
- Complete Blood Count