Calcium for Cardiovascular Health
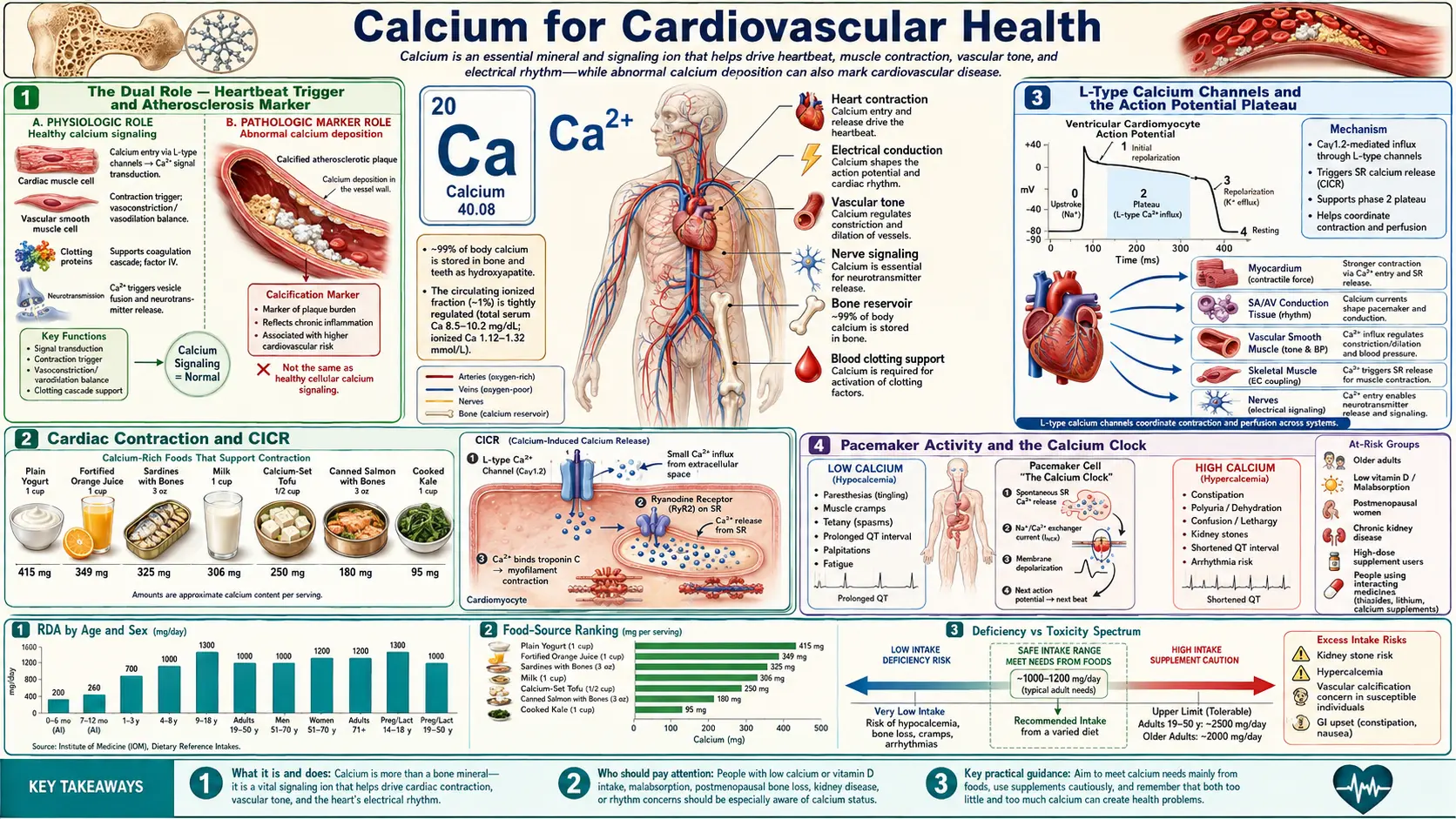
Calcium occupies an uncomfortable dual position in cardiovascular health. On one hand, transient Ca2+ flux is the universal trigger of every cardiac contraction — without calcium-induced calcium release (CICR) through L-type channels and ryanodine receptors, the heart would not beat. On the other hand, ectopic calcium deposition in coronary arteries and aortic valves is the structural signature of advanced atherosclerosis, and one of the strongest predictors of myocardial infarction. The 2010-2011 Bolland meta-analyses raised the unsettling possibility that high-dose isolated calcium supplementation might modestly increase cardiovascular event risk — a finding that reshaped osteoporosis prescribing practice worldwide. This page walks through the cellular electrophysiology that makes calcium indispensable to the beating heart, the vitamin K2 / Matrix Gla Protein axis that decides whether dietary calcium ends up in bone or in arterial walls, the calcium supplement controversy in detail, and the calcium channel blocker drug class as a pharmacological counterpoint that demonstrates the consequences of blocking L-type calcium entry into vascular smooth muscle.
Table of Contents
- The Dual Role — Heartbeat Trigger and Atherosclerosis Marker
- Cardiac Contraction and CICR
- L-Type Calcium Channels and the Action Potential Plateau
- Pacemaker Activity and the Calcium Clock
- Vascular Smooth Muscle Tone and Blood Pressure
- Vascular Calcification — The Vitamin K2 Axis
- The Calcium Supplement Controversy (Bolland)
- Calcium Channel Blockers — A Pharmacology Counterpoint
- Coronary Artery Calcium (CAC) Scoring
- Practical Recommendations
- Key Research Papers
- Connections
- Featured Videos
The Dual Role — Heartbeat Trigger and Atherosclerosis Marker
Calcium is unique among nutrients in that it is simultaneously essential for cardiovascular function and a marker of cardiovascular disease. Every cardiac contraction depends on a precisely choreographed rise and fall in intracellular calcium concentration that proceeds 60-100 times per minute for an entire lifetime. The same molecule, when deposited ectopically in arterial walls or heart valves, becomes a structural marker of atherosclerosis that can be quantified on a non-contrast CT scan and used as one of the most powerful independent predictors of future myocardial infarction.
This duality creates a complicated clinical picture. Patients with osteoporosis are often prescribed calcium supplements; those same patients tend to be older adults with significant cardiovascular risk. If calcium supplementation accelerates vascular calcification, the bone benefit comes at a cardiovascular cost. Resolving this trade-off requires understanding that the same calcium ion behaves very differently depending on where it ends up, and that nutrient partners (vitamin D3, vitamin K2, magnesium) heavily influence its final destination.
The rest of this page disentangles the cardiovascular calcium story into its constituent parts: the cellular mechanisms that make calcium indispensable to the heartbeat, the molecular biology that determines whether dietary calcium is deposited in bone or in arteries, the supplement-trial evidence that started the calcium-and-cardiovascular-events debate, and the pharmacological calcium channel blocker class that demonstrates what happens when calcium entry into vascular smooth muscle is therapeutically suppressed.
Cardiac Contraction and CICR
Cardiac muscle relies on a mechanism called calcium-induced calcium release (CICR). Unlike skeletal muscle, where the dihydropyridine receptor (DHPR) mechanically couples to and opens the ryanodine receptor (RyR1), in cardiac muscle the L-type DHPR opens during the action potential plateau and admits a small amount of extracellular calcium ("trigger calcium") into the cytoplasm. This trigger calcium then binds to RyR2 channels on the sarcoplasmic reticulum (SR) and amplifies into a much larger release of stored calcium into the cytoplasm.
- Trigger calcium fraction – The L-type calcium influx represents approximately 10% to 25% of the total cytoplasmic calcium that activates contraction; the remaining 75% to 90% comes from SR release through RyR2.
- Sarcoplasmic reticulum load – The amplitude of the calcium transient depends on the SR calcium content, which is determined by the balance between SERCA2a reuptake and ryanodine receptor leak. Beta-adrenergic stimulation increases SR load by phosphorylating phospholamban (releasing its inhibition of SERCA2a) and by phosphorylating L-type channels (increasing trigger calcium).
- Frank-Starling mechanism – When end-diastolic volume increases (greater stretch on the cardiac sarcomere), the myofilaments become more sensitive to a given concentration of calcium, increasing force of contraction without requiring a larger calcium transient. This intrinsic length-tension relationship matches cardiac output to venous return on a beat-to-beat basis.
- Calcium removal during diastole – For the heart to relax and fill during diastole, cytoplasmic calcium must be rapidly cleared. Approximately 70% is sequestered back into the SR by SERCA2a, 28% is extruded across the sarcolemma by the sodium-calcium exchanger (NCX), and the remaining 2% is handled by the plasma membrane calcium ATPase (PMCA) and mitochondrial uptake. Diastolic dysfunction (e.g., in HFpEF) often reflects impaired SERCA2a function and slower calcium reuptake.
- Arrhythmia from RyR2 leak – Diastolic SR calcium leak through dysfunctional RyR2 channels activates the inward NCX current, generating delayed afterdepolarizations (DADs) that can trigger ectopic beats and ventricular tachycardia. Gain-of-function RyR2 mutations cause catecholaminergic polymorphic ventricular tachycardia (CPVT), a stress-triggered lethal arrhythmia syndrome of childhood.
L-Type Calcium Channels and the Action Potential Plateau
The ventricular myocyte action potential has a distinctive square-wave shape with a prolonged plateau (phase 2) lasting roughly 200-300 milliseconds. This plateau is the defining electrophysiological feature of cardiac muscle and is created by inward L-type calcium current balanced against delayed-rectifier potassium currents. Three consequences flow from this plateau:
- It powers CICR – Sustained L-type calcium entry during the plateau provides the trigger calcium that opens RyR2 channels on the SR.
- It creates a long refractory period – Because sodium channels remain inactivated throughout the plateau, the cardiac myocyte cannot re-fire until repolarization is complete. This prevents tetanic contraction (sustained, uninterrupted force) of the heart muscle — which would be incompatible with diastolic filling and rapid death.
- It synchronizes contraction across the ventricle – The long plateau gives the activation wavefront time to traverse the entire ventricle, so that all the working myocardium contracts roughly together rather than as a peristaltic wave.
The L-type calcium channel (CaV1.2 in heart and vascular smooth muscle) is composed of a pore-forming alpha-1c subunit plus regulatory alpha-2/delta, beta, and gamma subunits. It is the target of the dihydropyridine calcium channel blocker class (amlodipine, nifedipine, felodipine) which preferentially blocks vascular smooth muscle channels, and the non-dihydropyridine class (verapamil, diltiazem) which preferentially blocks cardiac channels. These drugs are discussed below under the Calcium Channel Blockers section.
Pacemaker Activity and the Calcium Clock
The sinoatrial (SA) node is the heart's natural pacemaker. Unlike working ventricular myocytes, SA nodal cells have no stable resting membrane potential; they exhibit spontaneous diastolic depolarization that brings the cell to threshold and fires the next action potential. Two complementary mechanisms drive this automaticity:
- The "membrane clock" – The funny current (If) is a slowly activating mixed sodium-potassium inward current carried by HCN4 channels. It begins to flow as the cell repolarizes, slowly depolarizing the membrane. T-type calcium channels (CaV3) activate at intermediate voltages and contribute further to depolarization. Finally, L-type channels open and produce the action potential upstroke.
- The "calcium clock" – Rhythmic spontaneous calcium release events ("local calcium releases" or LCRs) from the SR through RyR2 channels occur during late diastole. These calcium releases activate the inward sodium-calcium exchanger current (3 Na+ in for 1 Ca2+ out is electrogenic, depolarizing the membrane), accelerating the pacemaker depolarization. The membrane clock and the calcium clock are tightly coupled and reinforce each other.
- Beta-adrenergic acceleration – Sympathetic stimulation increases heart rate by accelerating both clocks. PKA phosphorylation enhances If, increases L-type calcium current, and increases SR calcium load (more spontaneous calcium releases per diastolic interval). Parasympathetic stimulation has the opposite effect via M2 muscarinic receptor activation of inward-rectifier potassium channels.
- Sick sinus syndrome and ivabradine – Dysfunction of the funny current can produce inappropriate sinus bradycardia; the drug ivabradine selectively inhibits If and is used to lower heart rate in chronic heart failure and angina without the negative inotropic effects of beta-blockers.
Vascular Smooth Muscle Tone and Blood Pressure
Vascular smooth muscle cells in the walls of arteries and arterioles maintain partial contraction (tone) at rest. The intracellular calcium concentration in these cells is a primary determinant of vascular resistance and therefore of blood pressure.
- Calcium influx pathways – Vascular smooth muscle cells acquire calcium from extracellular fluid primarily through L-type calcium channels (CaV1.2). Membrane depolarization (e.g., from stretch, alpha-1 adrenergic stimulation, or angiotensin II signaling) opens these channels. Receptor-operated channels and store-operated channels (Orai/STIM) contribute additional calcium entry during sustained agonist stimulation.
- SR calcium release – Vasoconstrictor agonists that signal through Gq-coupled receptors (alpha-1 adrenergic, angiotensin II AT1, endothelin ETA, vasopressin V1a) activate phospholipase C, generating IP3 that opens IP3 receptors on the SR and releases stored calcium.
- Calmodulin-MLCK signaling – Cytoplasmic Ca2+ binds calmodulin, activating myosin light-chain kinase (MLCK), which phosphorylates the regulatory light chain of smooth muscle myosin. Phosphorylated myosin engages cross-bridge cycling with actin, generating force and constricting the vessel.
- Endothelial calcium and nitric oxide – In the endothelial cell layer that lines blood vessels, calcium signaling activates endothelial nitric oxide synthase (eNOS), generating nitric oxide that diffuses into adjacent smooth muscle, activates guanylyl cyclase, raises cGMP, and promotes vasodilation. This is the basis of endothelium-dependent dilation tested clinically by flow-mediated dilation (FMD).
- RhoA / Rho-kinase calcium sensitization – Beyond calcium concentration, vasoconstrictor signaling also activates the RhoA / Rho-kinase pathway, which inhibits myosin light-chain phosphatase (MLCP). This increases the force generated at any given calcium concentration. Fasudil (a Rho-kinase inhibitor used in cerebral vasospasm) acts on this pathway.
Vascular Calcification — The Vitamin K2 Axis
Vascular calcification is the deposition of calcium phosphate mineral (hydroxyapatite) within the wall of arteries. It comes in two anatomically and pathophysiologically distinct flavors: intimal calcification within atherosclerotic plaque (associated with classical cardiovascular risk factors) and medial calcification within the elastic lamina (Monckeberg's medial calcific sclerosis, more common in diabetes and chronic kidney disease, producing stiff but non-occlusive arteries).
What was once thought to be a passive precipitation reaction is now understood to be a tightly regulated, actively suppressed process. Vascular smooth muscle cells continuously synthesize Matrix Gla Protein (MGP), a small calcification-inhibiting protein that requires gamma-carboxylation of three glutamate residues to be functional. The gamma-carboxylation reaction requires vitamin K2 as a cofactor. When K2 is deficient, MGP remains uncarboxylated and cannot inhibit calcification — circulating uncarboxylated MGP (ucMGP) is a marker of vitamin K insufficiency and an independent risk factor for vascular calcification and cardiovascular events.
- The vitamin K1 vs K2 distinction – K1 (phylloquinone, from leafy greens) is preferentially taken up by the liver and used for coagulation factor carboxylation. K2 (menaquinone-7 from natto, MK-4 from animal foods) has wider tissue distribution and is the form that activates extra-hepatic Gla proteins including MGP and osteocalcin. The Rotterdam Study found that dietary K2 (but not K1) was associated with reduced coronary artery calcium, all-cause mortality, and cardiovascular mortality.
- Osteocalcin and the bone-vascular link – Osteocalcin is another vitamin-K-dependent Gla protein, secreted by osteoblasts. Carboxylated osteocalcin binds hydroxyapatite and helps direct calcium into bone matrix. Uncarboxylated osteocalcin reflects vitamin K insufficiency and is associated with both lower bone density and higher vascular calcification — the same molecular insufficiency drives both.
- Vitamin D context – Vitamin D increases intestinal calcium absorption; whether the absorbed calcium ends up in bone or in arteries depends in part on vitamin K2 sufficiency. The "calcium paradox" of supplementing D and Ca without K is a plausible mechanism behind the Bolland supplement-trial findings.
- Magnesium's protective role – Magnesium competes with calcium at several molecular sites and at the level of crystal nucleation. Higher serum and dietary magnesium is associated with lower vascular calcification in observational studies, plausibly because Mg2+ physically interferes with hydroxyapatite crystal growth in vascular tissue.
- Phosphate burden in CKD – Vascular calcification is markedly accelerated in chronic kidney disease, driven largely by high serum phosphate, elevated FGF23, and impaired vitamin K activity. This is why CKD patients have such high cardiovascular event rates — their arteries actively calcify in a fundamentally different process from classical atherosclerosis.
The Calcium Supplement Controversy (Bolland)
In 2010, Mark Bolland and colleagues at the University of Auckland published a meta-analysis in BMJ reporting a modest but statistically significant increase in myocardial infarction risk (approximately 27% relative increase) in trials of calcium supplements without vitamin D. A 2011 follow-up reanalysis of the Women's Health Initiative (WHI) Calcium / Vitamin D Supplementation Study limited dataset, combined with other trials, found similar signals. These papers caused a major reassessment of routine high-dose calcium supplementation in osteoporosis prevention.
- Magnitude and absolute risk – The relative risk increase was modest; in absolute terms, a few extra cardiovascular events per 1,000 person-years. For a single patient at low cardiovascular risk, the absolute effect is small.
- Plausible mechanism – A single bolus dose of calcium (e.g., 1,000 mg of calcium carbonate) produces a transient spike in serum ionized calcium that does not occur with dietary calcium spread across many meals. This spike could plausibly accelerate vascular calcification, especially in the setting of suboptimal vitamin K2 status.
- Dietary calcium is not implicated – The cardiovascular signal applies to supplemental calcium consumed as isolated boluses; observational data on dietary calcium consistently show neutral or favorable cardiovascular associations. Food matrices, the slower absorption kinetics from food, and the package of other nutrients (vitamin K2, magnesium, potassium) that come with calcium-rich foods may explain the difference.
- Counterarguments – The Bolland meta-analyses have been criticized for relying on post-hoc reanalysis of trials not originally designed to assess cardiovascular outcomes, for issues with the WHI dataset specifically, and for combining trials with heterogeneous calcium doses and durations. Other meta-analyses have not consistently reproduced the signal. The 2017 Reid review concluded the cardiovascular signal is real but modest.
- Clinical translation – Most osteoporosis guidelines now favor obtaining calcium from food when possible, and reserving calcium supplementation for patients with documented dietary insufficiency. When supplementation is used, dose splitting (500 mg with morning and evening meals rather than 1,000 mg once) reduces the calcium spike, and concurrent vitamin K2 (MK-7, 90-180 mcg/day) provides a plausible mechanism for directing the calcium to bone rather than arteries.
For broader context on cardiovascular risk reduction, see our Atherosclerosis page and our Vitamin K2 page.
Calcium Channel Blockers — A Pharmacology Counterpoint
The calcium channel blocker (CCB) drug class provides a useful counterpoint that illustrates what happens when calcium entry into vascular smooth muscle and cardiac myocytes is therapeutically suppressed.
- Dihydropyridines (DHPs) – Amlodipine, nifedipine, felodipine, nicardipine. These bind preferentially to L-type CaV1.2 channels in vascular smooth muscle, causing peripheral vasodilation and lowering blood pressure. They have minimal direct cardiac effects at therapeutic doses, though reflex sympathetic activation can produce tachycardia.
- Non-dihydropyridines (non-DHPs) – Verapamil and diltiazem. These act on L-type channels in cardiac myocytes and SA/AV nodal cells, producing negative inotropic and negative chronotropic effects in addition to vasodilation. They are used in rate control of atrial fibrillation and for symptomatic supraventricular tachycardia.
- Use in angina – CCBs reduce myocardial oxygen demand by lowering afterload (peripheral vasodilation), reduce heart rate (non-DHPs), and produce coronary vasodilation, all useful in chronic stable angina and vasospastic (Prinzmetal) angina.
- Use in hypertension – Amlodipine is one of the most widely prescribed antihypertensive drugs globally, used as first-line therapy especially in older adults and Black patients per JNC and ACC/AHA guidelines. The ALLHAT trial established the long-term efficacy of CCB-based antihypertensive therapy.
- Hemiplegic migraine and other channelopathies – Mutations in the neuronal P/Q-type calcium channel (CaV2.1, CACNA1A gene) cause familial hemiplegic migraine type 1, illustrating that calcium channels have non-cardiovascular roles in neuronal excitability.
- Side effects – Peripheral edema (especially DHPs, due to precapillary arteriolar dilation without postcapillary venular dilation), constipation (especially verapamil), gingival hyperplasia (rare), and bradycardia/AV block (non-DHPs).
- Hypocalcemia is not produced by CCBs – CCBs block voltage-gated channels in specific tissues; they do not lower serum total calcium concentration. Total body calcium balance is governed by the gut/kidney/bone axis and the PTH/vitamin D/calcitonin hormonal system, none of which CCBs affect directly.
Coronary Artery Calcium (CAC) Scoring
Coronary artery calcium scoring (also called the Agatston score) is a non-contrast CT scan that quantifies calcified plaque in the coronary arteries. It is one of the strongest single non-invasive predictors of future cardiovascular events and has been incorporated into the ACC/AHA cardiovascular risk assessment guidelines for selected intermediate-risk patients.
- Score interpretation – A CAC score of 0 indicates no detectable calcified plaque and a very low 10-year cardiovascular event risk. Scores of 1-99 indicate mild calcification; 100-399 moderate; 400 or greater extensive. A score above 100 is generally considered an indication for statin therapy regardless of the traditional risk-factor-based 10-year ASCVD estimate.
- Power-of-zero – The "CAC zero" finding is particularly useful for de-escalation of preventive therapy in patients who otherwise look high risk on lipid-and-blood-pressure-based calculators. The MESA cohort and other longitudinal datasets show that CAC = 0 confers a 10-year cardiovascular event rate around 1-2%, compared with 10-15% for CAC > 100.
- Race-ethnic and sex variation – CAC accrues at different rates in different populations; Black patients tend to have lower CAC at any given level of cardiovascular risk than White patients, and women lag men by approximately 10 years. The interpretation of CAC scores must therefore be calibrated to demographics.
- Limitations – CAC measures calcified plaque, but unstable (soft, vulnerable) plaque is the cause of most acute coronary syndromes. A patient can have a low CAC score and still have substantial non-calcified plaque. CAC scoring complements but does not replace functional cardiovascular testing in symptomatic patients.
- Indication and cost – Insurance coverage in the US is variable but expanding; out-of-pocket cost is typically $100-200. Radiation exposure is approximately 1 mSv, comparable to a mammogram.
Practical Recommendations
- Food first – Aim for 1,000-1,200 mg/day of calcium from dietary sources before considering supplementation. Dairy (milk, yogurt, cheese), calcium-set tofu, canned sardines and salmon with bones, leafy greens (kale, bok choy — not spinach due to oxalate), and almonds are the most calcium-dense foods.
- If supplementing – Use the smallest dose required to bridge the gap between dietary intake and target. Split doses to no more than 500 mg per administration, with meals. Pair with vitamin K2 (MK-7, 90-180 mcg/day) and adequate vitamin D3 (25(OH)D level >30 ng/mL).
- Form selection – Calcium citrate is absorbed with or without gastric acid and is preferred for patients on PPIs or H2 blockers or with achlorhydria. Calcium carbonate requires gastric acid and should be taken with food; it is cheaper and more dense (40% elemental calcium vs ~21% for citrate).
- Magnesium balance – Most adults consume insufficient magnesium. Aim for 400-420 mg/day (men) or 310-320 mg/day (women) from food or, if needed, glycinate or citrate supplements.
- Consider CAC scoring – For intermediate-risk patients (10-year ASCVD risk 5-20%) deciding about statin therapy, CAC scoring can substantially refine the decision.
- Avoid mega-doses – Stay below the upper intake level (2,500 mg/day adults 19-50; 2,000 mg/day 51+). Most adults do not need any supplemental calcium beyond what diet provides; high-dose supplementation should be reserved for diagnosed insufficiency under physician guidance.
This content is provided for informational purposes only and does not constitute medical advice. The choice of whether to supplement calcium — and at what dose — should be made with a physician who can weigh individual cardiovascular and bone-health context.
Key Research Papers
- Bolland MJ, Avenell A, Baron JA, et al. (2010). Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ. — DOI: 10.1136/bmj.c3691
- Bolland MJ, Grey A, Avenell A, Gamble GD, Reid IR (2011). Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women's Health Initiative Limited Access Dataset and meta-analysis. BMJ. — DOI: 10.1136/bmj.d2040
- Reid IR, Bolland MJ (2020). Calcium and/or vitamin D supplementation for the prevention of fragility fractures: who needs it? Nutrients. — DOI: 10.3390/nu12041011
- Bers DM (2002). Cardiac excitation-contraction coupling. Nature. — DOI: 10.1038/415198a
- Eisner DA, Caldwell JL, Kistamas K, Trafford AW (2017). Calcium and excitation-contraction coupling in the heart. Circulation Research. — DOI: 10.1161/CIRCRESAHA.117.310230
- Geleijnse JM, Vermeer C, Grobbee DE, et al. (2004). Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: the Rotterdam Study. Journal of Nutrition. — DOI: 10.1093/jn/134.11.3100
- Schurgers LJ, Cranenburg EC, Vermeer C (2008). Matrix Gla-protein: the calcification inhibitor in need of vitamin K. Thrombosis and Haemostasis. — PubMed
- Beulens JW, Bots ML, Atsma F, et al. (2009). High dietary menaquinone intake is associated with reduced coronary calcification. Atherosclerosis. — DOI: 10.1016/j.atherosclerosis.2008.07.010
- Detrano R, Guerci AD, Carr JJ, et al. (2008). Coronary calcium as a predictor of coronary events in four racial or ethnic groups. NEJM. — DOI: 10.1056/NEJMoa072100
- Greenland P, Blaha MJ, Budoff MJ, Erbel R, Watson KE (2018). Coronary calcium score and cardiovascular risk. Journal of the American College of Cardiology. — DOI: 10.1016/j.jacc.2017.10.099
- Catterall WA (2011). Voltage-gated calcium channels. Cold Spring Harbor Perspectives in Biology. — DOI: 10.1101/cshperspect.a003947
- Lakatta EG, Maltsev VA, Vinogradova TM (2010). A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circulation Research. — DOI: 10.1161/CIRCRESAHA.109.206078
- Reid IR, Bristow SM, Bolland MJ (2015). Calcium supplements: benefits and risks. Journal of Internal Medicine. — DOI: 10.1111/joim.12394
PubMed Topic Searches
- PubMed: Calcium supplements / CV events
- PubMed: Vascular calcification / MGP / K2
- PubMed: CAC score / Agatston
- PubMed: Cardiac CICR / L-type
- PubMed: Calcium channel blockers / hypertension
- PubMed: Menaquinone / MK-7 / coronary calcium
External Authoritative Resources
- American Heart Association — Coronary Calcium Scan
- Linus Pauling Institute — Calcium
- NIH Office of Dietary Supplements — Calcium Fact Sheet
- ACC/AHA — 2018 Guideline on the Management of Blood Cholesterol (CAC scoring discussion)
Connections
- Calcium (Main Page)
- Calcium Benefits Hub
- Calcium for Bone Health
- Calcium for Muscle Function
- Calcium for Nerve Transmission
- Vitamin K2 (MGP Cofactor)
- Vitamin D3
- Magnesium
- Magnesium and Heart Health
- Potassium
- Atherosclerosis
- Hypertension
- Arrhythmia
- Stroke
- Osteoporosis
- Kidney Disease (CKD calcification)
- All Minerals