Atherosclerosis
Interactive Visualization The Heart & Circulation — watch a blood cell make the loop Trace a blood cell through all four chambers and both circuits, with a live ECG, chamber pressures, and an exercise mode. Launch → Interactive Visualization Cholesterol & the Artery Wall — watch a plaque form Follow LDL particles into the artery wall and see foam cells build plaque — then add a statin and watch the receptors clear them. Launch → Interactive Visualization Nitric Oxide — make an artery relax Watch the endothelium turn L-arginine into nitric oxide and widen the artery — then let oxidative stress uncouple eNOS and stiffen it, or pour in beet nitrate to rescue the flow. Launch → Interactive Visualization Bile Acids — emulsify a fatty meal yourself Eat a fatty meal and watch bile squirt from the gallbladder to emulsify the fat into droplets lipase can attack — then block the bile and watch the fat, and vitamins A, D, E and K, go straight through. Launch →
Table of Contents
- What is Atherosclerosis?
- Stages of Plaque Progression
- Endothelial Dysfunction
- Risk Factors
- Diagnosis
- Prevention and Treatment
- Complications
- Research Papers
- Connections
- Featured Videos
What is Atherosclerosis?
Atherosclerosis is a chronic, progressive disease in which fatty deposits, cholesterol, calcium, and inflammatory cells accumulate inside the walls of medium and large arteries, forming plaques that narrow the lumen and stiffen the vessel. The term comes from the Greek athere (gruel, referring to the soft lipid-rich core of the plaque) and sklerosis (hardening, referring to the fibrous cap that overlies it).
It is the underlying cause of most heart attacks, ischemic strokes, peripheral artery disease, and many cases of sudden cardiac death. Cardiovascular disease driven by atherosclerosis is the leading cause of death worldwide, accounting for roughly 18–20 million deaths annually. In the United States alone, more than half of adults over age 40 have measurable arterial plaque on imaging, even when they feel completely well.
The disease begins silently — often in childhood — and progresses over decades. Fatty streaks have been documented in the arteries of teenagers and young adults autopsied after accidental death. Most people remain asymptomatic until a plaque has narrowed an artery by more than 70%, or until a previously stable plaque ruptures and triggers a clot. By the time the first symptom appears, the disease is typically advanced and systemic, affecting multiple arterial beds simultaneously.
Atherosclerosis is no longer viewed as simple "cholesterol clogging the pipes." Modern understanding treats it as a chronic inflammatory disease of the arterial wall, driven by the retention and oxidation of apolipoprotein B-containing lipoproteins (LDL, VLDL remnants, and Lp(a)) inside the subendothelial space, with secondary recruitment of immune cells and progressive remodeling of the vessel.
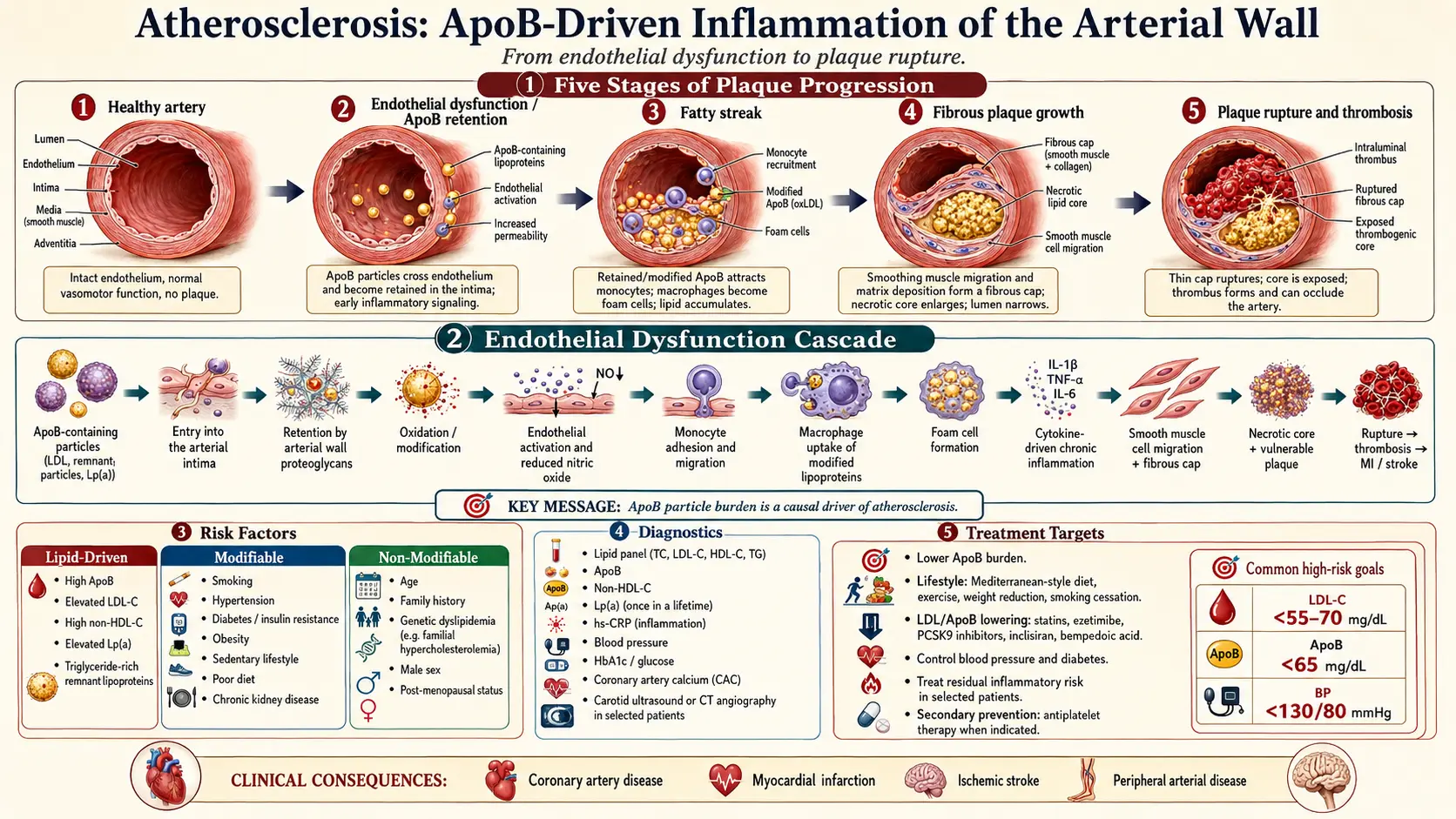
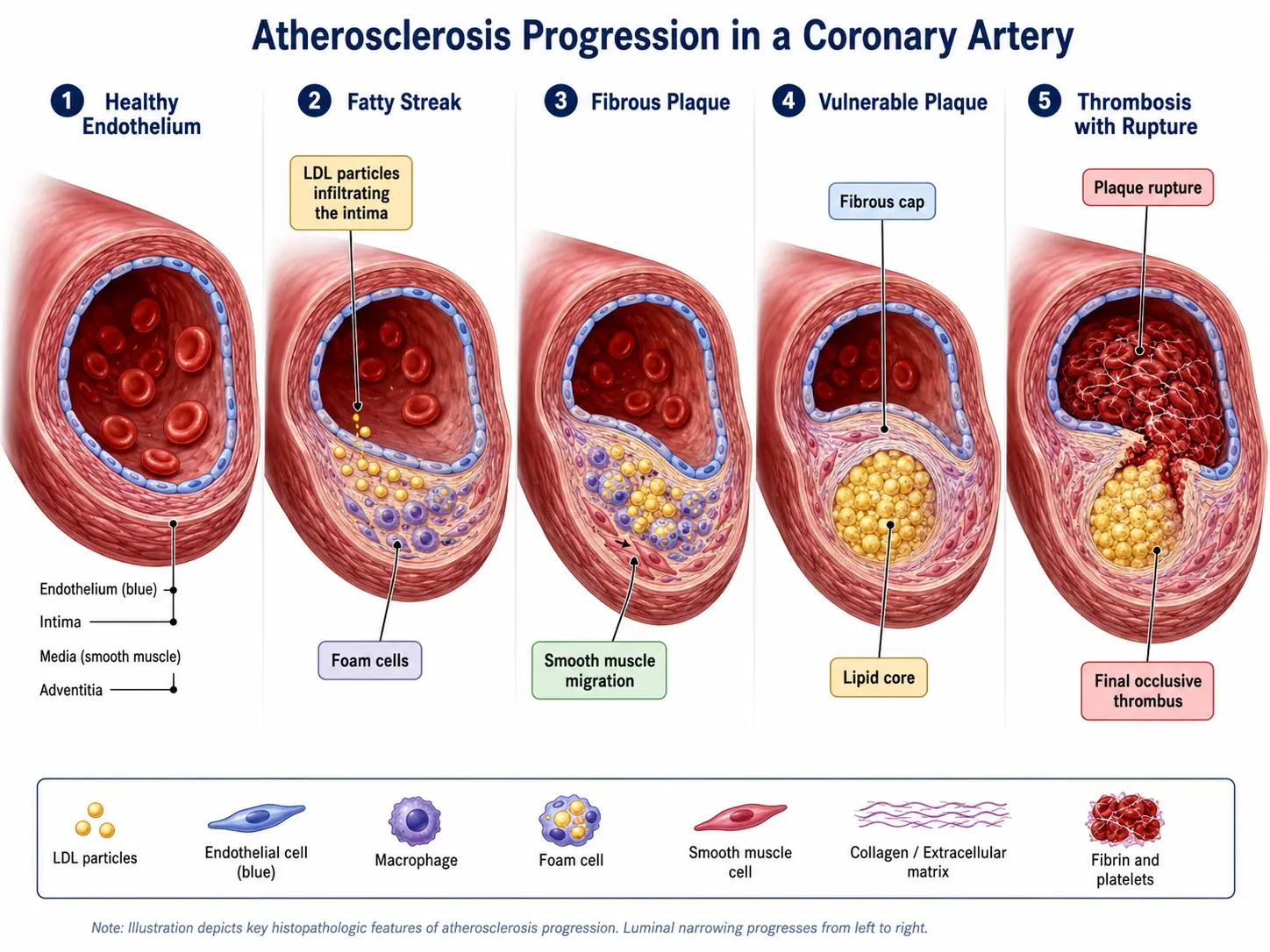
Stages of Plaque Progression
Atherosclerosis progresses through a recognized sequence of histologic stages. The illustration above traces five of these stages, from a healthy artery to a ruptured plaque with overlying thrombus.
Stage 1: Healthy Artery
A normal artery has three layers — the inner intima lined by a single sheet of endothelial cells, the muscular media, and the outer adventitia. The endothelium is a metabolically active barrier that produces nitric oxide (NO), regulates vascular tone, prevents platelet aggregation, and selectively controls what crosses into the vessel wall.
Stage 2: Fatty Streak
The earliest visible lesion is a fatty streak — a flat, yellowish patch on the inner surface of the artery. It forms when LDL particles cross a dysfunctional endothelium and become trapped in the intima, where they undergo oxidation. Monocytes follow them in, differentiate into macrophages, engulf the oxidized LDL, and become lipid-laden foam cells. Fatty streaks themselves do not block blood flow and can theoretically regress, but they are the seed of every later stage.
Stage 3: Fibrous (Stable) Plaque
If the inflammatory stimulus persists, smooth muscle cells migrate from the media into the intima, proliferate, and lay down collagen, forming a thick fibrous cap over a growing lipid core. This is the classical "stable" or fibroatheromatous plaque. It can grow large enough to narrow the lumen and produce angina with exertion (when oxygen demand exceeds the restricted supply), but the cap holds and no clot forms.
Stage 4: Vulnerable (Unstable) Plaque
A subset of plaques become vulnerable — characterized by a large lipid core, a thin fibrous cap (often less than 65 micrometers thick), heavy macrophage infiltration, and active inflammation that secretes matrix metalloproteinases. These enzymes digest the collagen of the cap from below. Vulnerable plaques are often not the largest plaques and frequently do not produce symptoms before they rupture — which is why a "passing" stress test does not rule out future heart attack.
Stage 5: Plaque Rupture and Thrombosis
When the thin cap finally tears, the highly thrombogenic lipid core and tissue factor are exposed to flowing blood. Platelets aggregate, the coagulation cascade ignites, and a thrombus forms within minutes. If the clot fully occludes the artery, the downstream tissue infarcts — a myocardial infarction in the coronary arteries, an ischemic stroke in the cerebral circulation, or acute limb ischemia in the legs. Most acute coronary events occur on plaques that previously caused less than 50% stenosis, which is why population-level prevention (lowering ApoB lifelong) outperforms reactive stenting of the worst-looking lesion.
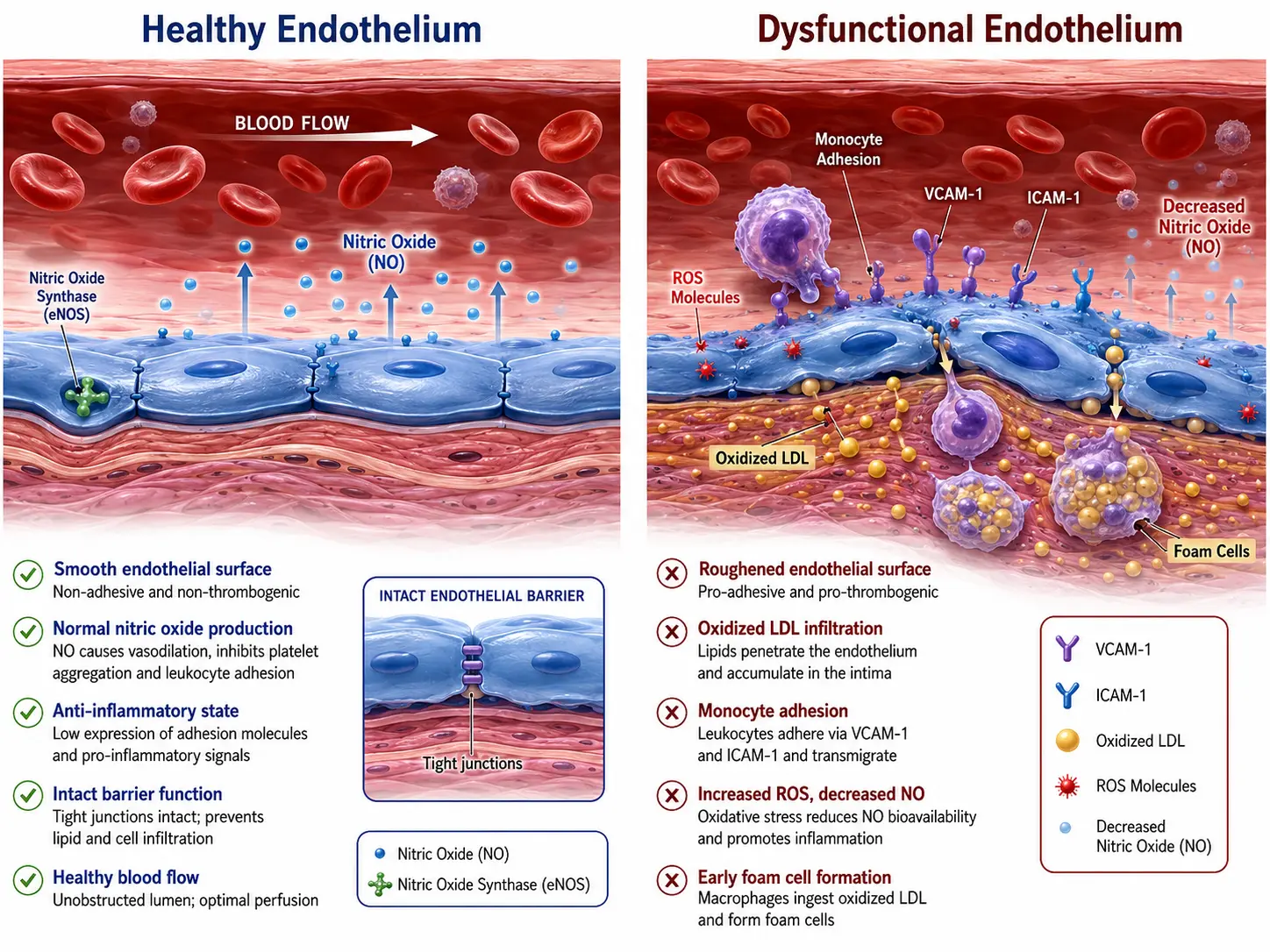
Endothelial Dysfunction
Endothelial dysfunction is the initiating event in atherosclerosis. Long before any plaque is visible on imaging, the endothelium has already shifted from a protective, anti-inflammatory state to a pro-inflammatory, pro-thrombotic one. The illustration above shows the cellular machinery driving that shift.
Loss of Nitric Oxide
A healthy endothelium continuously produces nitric oxide (NO) via the enzyme endothelial NO synthase (eNOS). NO relaxes the underlying smooth muscle, inhibits platelet adhesion, and suppresses inflammatory gene expression. Risk factors such as high LDL, high blood pressure, hyperglycemia, and smoking generate reactive oxygen species (ROS) that scavenge NO and uncouple eNOS, so the enzyme begins producing superoxide instead of NO. The vessel loses its ability to dilate, and antioxidant capacity collapses.
LDL Retention and Oxidation
Apolipoprotein B-containing lipoproteins (LDL, VLDL remnants, Lp(a)) cross the dysfunctional endothelium and bind to proteoglycans in the intima. Trapped LDL particles are progressively oxidized by ROS into oxidized LDL (oxLDL). Native LDL is largely inert, but oxLDL is intensely pro-inflammatory: it activates endothelial adhesion molecules (VCAM-1, ICAM-1, E-selectin) and chemokines (MCP-1) that recruit circulating monocytes.
Monocyte Adhesion and Foam Cell Formation
Monocytes roll along the activated endothelium, adhere to it, and squeeze between the cells into the intima. There they differentiate into macrophages and dramatically upregulate scavenger receptors (CD36, SR-A, LOX-1) that pull oxLDL into the cell unchecked — unlike the native LDL receptor, scavenger receptors are not feedback-suppressed by intracellular cholesterol. The macrophages fill with cholesterol esters, take on a foamy appearance under the microscope, and become foam cells. Foam cells secrete more inflammatory cytokines (TNF-α, IL-1β, IL-6), recruiting yet more monocytes and amplifying the lesion.
Why It Matters
Endothelial dysfunction is detectable years before any plaque forms, using techniques such as flow-mediated dilation of the brachial artery. Lifestyle interventions (exercise, Mediterranean diet, smoking cessation) and pharmacologic therapy (statins, ACE inhibitors) can measurably restore endothelial function. Lowering ApoB-containing lipoproteins removes the substrate that the dysfunctional endothelium retains, which is why aggressive LDL-lowering arrests — and in some imaging trials reverses — plaque growth.
Risk Factors
Atherosclerosis risk is driven by a small number of dominant factors. The first three are causal in the strongest sense: lifelong exposure to elevated levels independently produces the disease.
Causal (Lipid-Driven) Factors
- LDL cholesterol and ApoB: Every ApoB-containing particle (LDL, VLDL remnants, IDL, Lp(a)) carries a single ApoB molecule, so ApoB is a one-to-one count of atherogenic particles and is a more accurate risk marker than LDL-C in patients with metabolic syndrome, diabetes, or high triglycerides. Mendelian randomization studies show that lifelong low LDL/ApoB — whether from genetics or pharmacology — produces a much larger reduction in cardiovascular events than late-life lowering of the same magnitude.
- Lipoprotein(a) — Lp(a): An LDL-like particle with an attached apolipoprotein(a) tail. Levels are 80–90% genetic and largely lifelong-fixed. Roughly 1 in 5 adults has elevated Lp(a) (above 50 mg/dL or 125 nmol/L), which independently raises risk of MI, stroke, and aortic stenosis. Should be measured at least once in every adult.
- Hypertension: Mechanically stresses the endothelium, particularly at branch points and curvatures where shear stress is already disturbed, accelerating endothelial dysfunction and plaque development.
Major Modifiable Factors
- Type 2 diabetes and insulin resistance: Hyperglycemia glycates LDL (making it more atherogenic), drives oxidative stress, and produces a pro-inflammatory state. Diabetes roughly doubles cardiovascular risk and is considered a coronary heart disease "risk equivalent."
- Smoking: Damages endothelium directly, raises ROS, lowers HDL, and dramatically increases thrombotic risk on top of any underlying plaque burden. Smokers have higher rates of plaque rupture at any given plaque size.
- Inflammation (hsCRP): A high-sensitivity C-reactive protein level above 2 mg/L identifies "residual inflammatory risk" that persists even after LDL is well controlled. The CANTOS trial confirmed that targeting inflammation directly reduces events.
- Sedentary lifestyle: Independent of weight, low physical activity worsens endothelial function, lipid profile, blood pressure, and insulin sensitivity.
- Diet: Diets high in industrially processed foods, refined carbohydrates, and trans fats raise ApoB and inflammation; the Mediterranean and DASH patterns lower it.
- Visceral obesity: Abdominal fat is metabolically active and drives insulin resistance, dyslipidemia, and a pro-inflammatory state.
Non-Modifiable Factors
- Age: Plaque burden is essentially cumulative exposure to ApoB over time, so risk rises sharply with age in both sexes.
- Sex: Men develop clinically apparent disease 7–10 years earlier on average; women catch up after menopause.
- Family history: Premature CAD in a first-degree relative (men under 55, women under 65) is a strong independent risk marker. Familial hypercholesterolemia and elevated Lp(a) often run in families.
Diagnosis
Because atherosclerosis is silent until late, the goal of modern cardiology is to detect and quantify subclinical disease before it ruptures. Several complementary tools are used.
Blood Tests
- Standard lipid panel: Total cholesterol, LDL-C, HDL-C, triglycerides. Inexpensive, universal, but LDL-C can be misleading when triglycerides are high or particles are small and dense.
- ApoB: A direct count of all atherogenic particles. Increasingly considered the preferred lipid risk marker, especially in metabolic syndrome and diabetes. Goal in primary prevention is generally below 90 mg/dL; in secondary prevention, below 60–65 mg/dL.
- Lipoprotein(a): Should be measured at least once in adulthood. Levels are largely genetic and stable across life. Above 50 mg/dL (125 nmol/L) is high-risk.
- hsCRP: High-sensitivity C-reactive protein, a marker of systemic inflammation. Above 2 mg/L marks elevated residual inflammatory risk.
- HbA1c and fasting glucose: Screen for diabetes and prediabetes.
Imaging
- Carotid intima-media thickness (CIMT): Ultrasound measurement of the inner two layers of the carotid artery. A thicker CIMT correlates with overall atherosclerotic burden. Inexpensive, radiation-free, but operator-dependent.
- Coronary artery calcium (CAC) score: A non-contrast CT of the heart that quantifies calcified plaque using the Agatston score. A score of 0 in a middle-aged adult is one of the strongest negative predictors in medicine ("the power of zero"). A score above 100 places a patient in a high-risk category regardless of LDL number; above 400 is very high risk. CAC is the single most powerful tool for refining risk in the intermediate-risk patient.
- Coronary CT angiography (CCTA): A contrast-enhanced CT that visualizes the lumen and the wall, including non-calcified soft plaque that CAC misses. Increasingly used as a first-line non-invasive test for chest pain (the SCOT-HEART and PROMISE trials).
- Stress testing (exercise ECG, stress echo, nuclear MPI): Detects flow-limiting stenosis (typically >70%) but is insensitive to early plaque, so a normal stress test does not rule out atherosclerosis.
- Invasive coronary angiography: The historical gold standard for lumen narrowing. Reserved for patients with strong evidence of obstructive disease or planned intervention.
Prevention and Treatment
Treatment has two coupled goals: lower ApoB lifelong to remove the substrate of plaque growth, and address every modifiable risk factor to keep the endothelium healthy and inflammation low.
Lifestyle Foundation
- Mediterranean dietary pattern: Vegetables, fruits, legumes, nuts, whole grains, fish, and extra-virgin olive oil. The PREDIMED trial showed roughly a 30% reduction in major cardiovascular events versus a low-fat control diet in high-risk adults.
- Physical activity: At least 150 minutes per week of moderate aerobic activity, plus two strength sessions. Independently improves endothelial function, lipid profile, blood pressure, and insulin sensitivity.
- Smoking cessation: The single highest-impact behavior change. Cardiovascular risk drops measurably within months and approaches that of never-smokers within 10–15 years.
- Sleep and stress: 7–9 hours of sleep per night and active stress management lower blood pressure, inflammation, and atherogenic dyslipidemia.
- Weight and waist: Reducing visceral adiposity improves every component of metabolic syndrome.
Lipid-Lowering Medications
- Statins (atorvastatin, rosuvastatin, simvastatin, pravastatin): The first-line therapy. Inhibit hepatic HMG-CoA reductase, upregulate LDL receptors, and lower LDL-C by 30–55%. Decades of trials (4S, HPS, JUPITER, IMPROVE-IT, etc.) show consistent reductions in MI, stroke, and cardiovascular death of roughly 25% per 1 mmol/L (39 mg/dL) reduction in LDL.
- Ezetimibe: Blocks intestinal cholesterol absorption via NPC1L1, lowers LDL by an additional 15–20% on top of a statin, and reduces events (IMPROVE-IT).
- PCSK9 inhibitors (evolocumab, alirocumab): Monoclonal antibodies that prolong LDL receptor lifespan, lowering LDL by an additional 50–60% on top of statin + ezetimibe. The FOURIER and ODYSSEY OUTCOMES trials showed further event reductions in patients with established disease. Inclisiran is a small interfering RNA with similar potency dosed twice yearly.
- Bempedoic acid: An ATP-citrate lyase inhibitor active in the liver but not muscle, useful in statin-intolerant patients. The CLEAR Outcomes trial (2023) confirmed event reduction.
- Icosapent ethyl (high-dose EPA): The REDUCE-IT trial showed a 25% reduction in major events in statin-treated patients with elevated triglycerides.
Antiplatelet and Adjunctive Therapy
- Aspirin: Indicated for secondary prevention (after MI, stroke, stent, peripheral disease). For primary prevention, the bleeding risk often outweighs benefit and current guidelines reserve it for selected high-risk patients.
- Blood pressure control: Generally to below 130/80 mm Hg in adults with cardiovascular risk; ACE inhibitors, ARBs, calcium channel blockers, and thiazide diuretics are first line.
- Glycemic control with cardiovascular benefit: SGLT2 inhibitors (empagliflozin, dapagliflozin) and GLP-1 receptor agonists (semaglutide, liraglutide) reduce cardiovascular events in diabetes and increasingly in selected non-diabetic populations.
- Targeted anti-inflammatory therapy: Low-dose colchicine (LoDoCo2, COLCOT) and the IL-1β antibody canakinumab (CANTOS) reduce events in patients with elevated hsCRP, validating inflammation as a therapeutic target.
Procedures for Established Disease
- Percutaneous coronary intervention (PCI): Stenting an obstructive lesion. Symptom-relieving for angina and lifesaving in acute MI; in stable disease (ISCHEMIA trial), it is generally not superior to optimal medical therapy for preventing future events.
- Coronary artery bypass grafting (CABG): Preferred over PCI for patients with extensive multi-vessel or left-main disease, particularly those with diabetes (FREEDOM trial).
- Carotid endarterectomy or stenting: For high-grade carotid stenosis with prior stroke or TIA.
Complications
Atherosclerosis is a systemic disease — a patient with plaque in one bed almost always has plaque in others. The clinical picture depends on which artery fails first.
- Myocardial infarction (heart attack): Plaque rupture in a coronary artery with overlying thrombus, occluding flow and causing irreversible heart muscle death. Roughly 800,000 occur in the United States per year.
- Stable angina and chronic coronary syndromes: Exertional chest pain from a flow-limiting (but not yet ruptured) coronary plaque.
- Ischemic stroke: Either embolic from a ruptured carotid plaque or local in-situ thrombosis of an intracranial artery. Atherosclerosis underlies the large-vessel and small-vessel subtypes; cardioembolic strokes (atrial fibrillation) are a separate mechanism that often coexists.
- Peripheral artery disease (PAD): Plaque in the iliac, femoral, and tibial arteries causes intermittent claudication (calf pain with walking that resolves with rest) and, if severe, critical limb ischemia and gangrene. PAD is a strong marker of generalized atherosclerosis and predicts a high cardiovascular event rate.
- Abdominal aortic aneurysm (AAA): Atherosclerosis weakens the aortic wall, allowing it to dilate. A ruptured AAA is rapidly fatal. One-time ultrasound screening is recommended for men aged 65–75 with a smoking history.
- Renal artery stenosis: Atherosclerotic narrowing of the renal arteries that can produce treatment-resistant hypertension and progressive kidney dysfunction.
- Mesenteric ischemia: Plaque in the celiac, superior mesenteric, or inferior mesenteric arteries can cause postprandial abdominal pain ("intestinal angina") and, when acute, life-threatening bowel infarction.
- Erectile dysfunction: Often the earliest symptom of generalized atherosclerosis in men, since the penile arteries are small and become flow-limited before larger beds.
- Vascular dementia and cognitive decline: Cerebral small-vessel disease and recurrent silent infarcts contribute substantially to age-related cognitive impairment.
Research Papers
The following PubMed topic searches return current peer-reviewed literature relevant to atherosclerosis. Each link opens a live PubMed query.
- Atherosclerosis pathophysiology
- Oxidized LDL endothelium
- Foam cells macrophages
- Vulnerable plaque rupture
- Statin clinical trials
- PCSK9 inhibitor outcomes
- ApoB cardiovascular risk
- Lp(a) atherosclerosis
- Coronary calcium score
- Intima-media thickness
- Inflammation hsCRP
- Mediterranean diet cardiovascular
Connections
- Cardiology
- Bile Acids: How You Digest Fat — interactive animation
- Nitric Oxide & Vessel Dilation — interactive animation
- Cholesterol & the Artery Wall — interactive animation
- The Heart & Circulation — interactive animation
- ApoB
- Coronary Calcium Score
- Lipid Panel
- Lipoprotein(a)
- Stroke
- Hypertension
- Cardiovascular Disease
- Coronary Artery Disease
- Cholesterol Management
- Peripheral Artery Disease
- Diabetes
- Metabolic Syndrome
- Insulin Resistance
- Anti-Inflammatory Diet
- Omega-3 Fatty Acids
- Berberine
- Garlic
- Magnesium
- Gum Disease