Calcium for Nerve Transmission
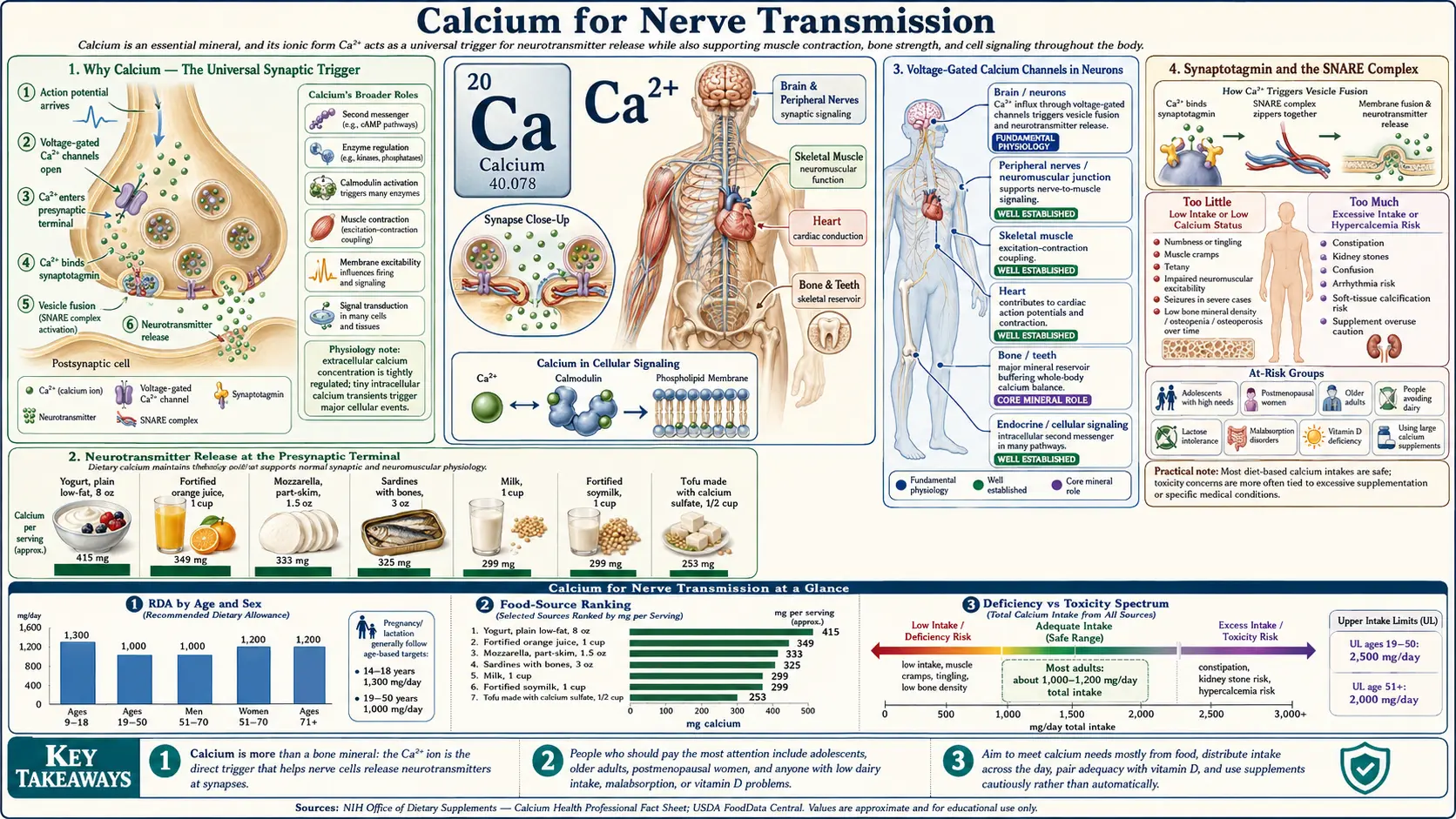
Calcium is the master molecule of neural communication. Every chemical synapse in the brain, every neuromuscular junction in the body, every neuroendocrine vesicle release, every long-term-potentiation event that lays down a memory — all begin with a presynaptic calcium spike. The voltage-gated calcium channels (P/Q-type, N-type) at presynaptic terminals open within microseconds of an arriving action potential, admitting a localized Ca2+ nanodomain that binds synaptotagmin and catalyzes SNARE-mediated vesicle fusion. The same molecule that builds bone and contracts muscle is the spark that turns electrical signals into chemical messages, and the chemical messages back into electrical signals in the next cell. Hypocalcemia bypasses this exquisite specificity and turns the entire peripheral nervous system hyperexcitable — producing the perioral tingling, carpopedal spasm (Trousseau), facial twitch (Chvostek), and in severe cases the laryngospasm and seizures that mark calcium-deficiency tetany. This page walks through the molecular machinery of synaptic transmission, the role of voltage-gated calcium channels in pacemaker neurons and action potential initiation, the calcium-induced calcium release that amplifies signals inside the neuron, the seizure-threshold consequences of hypocalcemia, and the channelopathies (Lambert-Eaton, familial hemiplegic migraine) that show what goes wrong when neuronal calcium signaling fails.
Table of Contents
- Why Calcium — The Universal Synaptic Trigger
- Neurotransmitter Release at the Presynaptic Terminal
- Voltage-Gated Calcium Channels in Neurons
- Synaptotagmin and the SNARE Complex
- Calcium-Induced Calcium Release in Neurons
- The Neuromuscular Junction
- Hypocalcemia — Chvostek, Trousseau, and Tetany
- Calcium and Seizure Threshold
- Calcium Channelopathies (LEMS, Hemiplegic Migraine, CPVT)
- Calcium Signaling in Aging and Cognition
- Key Research Papers
- Connections
- Featured Videos
Why Calcium — The Universal Synaptic Trigger
Of all the ions involved in cellular signaling, calcium occupies a unique position. Its intracellular free concentration is held at extraordinarily low levels at rest — approximately 100 nanomolar, compared with roughly 1.2 millimolar in extracellular fluid, a 10,000-fold gradient. Opening a voltage-gated calcium channel for a millisecond can produce a 100-fold local rise in cytoplasmic Ca2+ within nanometers of the channel mouth. This combination of (a) a massive electrochemical gradient, (b) high binding selectivity by sensor proteins, and (c) the rarity of free Ca2+ in the resting cytoplasm makes calcium a uniquely sharp signal: when it appears, the cell knows something specific just happened.
In neurons, this property is harnessed at multiple scales. At the millisecond scale, presynaptic calcium influx triggers neurotransmitter vesicle fusion. At the seconds-to-minutes scale, calcium activates CaMKII to produce long-term potentiation, the molecular substrate of memory. At the hours scale, calcium-dependent transcription factors (CREB, NFAT) regulate gene expression that supports synaptic plasticity. The same ion that signals "fire now" also signals "remember this."
The flip side of calcium's signal sharpness is the danger of calcium overload. Excessive cytoplasmic calcium activates proteases (calpains), nucleases, phospholipases, and the permeability transition pore in mitochondria, triggering apoptosis or necrosis. Glutamate excitotoxicity in ischemic stroke is fundamentally a calcium overload phenomenon: excessive NMDA receptor activation admits enough Ca2+ to overwhelm cellular buffers and execute the cell.
Neurotransmitter Release at the Presynaptic Terminal
The textbook sequence of chemical synaptic transmission is one of the most precisely characterized cell biological processes:
- Action potential arrival – A depolarizing wave reaches the presynaptic axon terminal, depolarizing the membrane from approximately −70 mV to around 0 to +30 mV.
- Voltage-gated calcium channel opening – P/Q-type (CaV2.1) and N-type (CaV2.2) channels in the active zone open within microseconds of depolarization. Each channel admits roughly 10,000-100,000 calcium ions per millisecond of opening.
- Calcium nanodomain formation – A localized calcium "cloud" forms within tens of nanometers of each open channel, reaching transient concentrations of 10-100 micromolar — a thousand-fold above resting levels — before being rapidly buffered and pumped away.
- Synaptotagmin calcium binding – Synaptotagmin-1, the principal fast calcium sensor on the synaptic vesicle, has two C2 domains (C2A and C2B) that bind Ca2+. Each C2 domain binds 3-5 calcium ions; the cooperative binding is what makes vesicle fusion a switch-like response to the calcium signal.
- SNARE complex zippering – Calcium-bound synaptotagmin triggers full zippering of the SNARE complex (vesicle-associated VAMP/synaptobrevin paired with target-membrane syntaxin-1 and SNAP-25). This brings the vesicle membrane into contact with the plasma membrane, driving fusion.
- Exocytosis and neurotransmitter release – The fusion pore opens, releasing the vesicle contents (~5,000 molecules of glutamate, GABA, ACh, or other transmitter) into the synaptic cleft. The released transmitter diffuses across the ~20-nanometer cleft and binds postsynaptic receptors.
- Endocytic recycling – Vesicle membrane is retrieved from the plasma membrane via clathrin-mediated endocytosis or kiss-and-run mechanisms, refilled with neurotransmitter via vesicular transporters, and re-docked at the active zone for the next round of release.
The entire sequence from action potential arrival to neurotransmitter release takes approximately 200 microseconds. This speed is critical for high-frequency synaptic transmission and for the millisecond-precision timing required for processes like sound localization in the auditory system.
Voltage-Gated Calcium Channels in Neurons
The voltage-gated calcium channel (VGCC) superfamily has ten members in mammals, grouped into three families distinguished by activation voltage, kinetics, and pharmacology. Each plays a distinct role in neural function:
- L-type (CaV1.2, CaV1.3) – "Long-lasting" high-voltage activated channels. In neurons they cluster in cell bodies and dendrites, where they contribute to plateau depolarizations, dendritic calcium spikes, and gene-expression coupling via the calcium-CaMKIV-CREB pathway. CaV1.3 has a relatively negative activation voltage and contributes to pacemaker activity in substantia nigra dopamine neurons — this is relevant to Parkinson's disease, where CaV1.3 calcium load may contribute to neuronal vulnerability.
- P/Q-type (CaV2.1) – The principal channel mediating fast synaptic transmission in the central nervous system and at the neuromuscular junction. Concentrated at presynaptic active zones. Mutations cause familial hemiplegic migraine type 1, episodic ataxia type 2, and spinocerebellar ataxia type 6. Blocked by omega-agatoxin from the funnel-web spider.
- N-type (CaV2.2) – "Neuronal" channels concentrated at peripheral and central nervous system presynaptic terminals. Important for pain signaling in the dorsal root ganglion / dorsal horn. The drug ziconotide (a synthetic version of omega-conotoxin MVIIA from the marine cone snail) selectively blocks N-type channels and is used intrathecally for severe chronic pain.
- R-type (CaV2.3) – "Residual" channels with roles in dendritic calcium signaling, synaptic plasticity, and some forms of neurotransmitter release.
- T-type (CaV3.1, CaV3.2, CaV3.3) – "Transient" low-voltage activated channels that activate near resting membrane potential and inactivate rapidly. They contribute to burst firing in thalamic relay neurons (relevant to thalamocortical oscillations and absence seizures), to pacemaker activity in some neurons, and to nociception in dorsal root ganglion neurons. The drug ethosuximide treats absence seizures by blocking thalamic T-type channels.
Selective pharmacology of these channel subtypes is a long-standing therapeutic frontier. Dihydropyridine calcium channel blockers (amlodipine, nifedipine) target L-type channels but with strong vascular smooth muscle selectivity, so they have minimal CNS effects at antihypertensive doses. Ziconotide (intrathecal) and ethosuximide (oral) demonstrate that selective neuronal calcium channel modulation is clinically achievable.
Synaptotagmin and the SNARE Complex
Synaptotagmin is the calcium sensor that converts a presynaptic calcium signal into vesicle fusion. The synaptotagmin family has at least 17 isoforms in mammals; synaptotagmin-1 (Syt1) is the principal fast calcium sensor at most CNS synapses, while synaptotagmin-2 dominates at fast-firing synapses such as the calyx of Held. Asynchronous release (the slow component of neurotransmitter release seen after high-frequency stimulation) is mediated by other isoforms with different calcium binding kinetics.
- Two C2 domains – Each synaptotagmin contains a transmembrane anchor and two cytoplasmic C2 domains (C2A and C2B). Each C2 domain binds 3-5 calcium ions in a coordination pocket formed by aspartate residues. The cooperative binding produces a steep dose-response: release probability rises with approximately the fourth power of calcium concentration.
- Phospholipid binding – Once calcium-loaded, the C2 domains insert into the inner leaflet of the plasma membrane, particularly into phosphatidylserine-rich regions. This brings the vesicle membrane into close apposition with the plasma membrane.
- SNARE interaction – Synaptotagmin also interacts directly with the assembled SNARE complex (VAMP/synaptobrevin + syntaxin-1 + SNAP-25), promoting full zippering and fusion.
- Complexin clamp – Complexin proteins bind partially assembled SNARE complexes and clamp them in a "ready-to-go" state, preventing premature fusion. Calcium-bound synaptotagmin displaces complexin and releases the clamp, allowing fusion. This clamping mechanism is what gives synaptic transmission its tight temporal precision.
- Botulinum and tetanus toxins – These bacterial neurotoxins are zinc-dependent proteases that cleave specific SNARE proteins (botulinum types A and E cleave SNAP-25; type B and tetanus cleave VAMP; type C cleaves syntaxin), abolishing vesicle fusion and producing flaccid paralysis (botulism) or spastic paralysis (tetanus, by selectively blocking inhibitory glycinergic transmission in the spinal cord).
Calcium-Induced Calcium Release in Neurons
Calcium-induced calcium release (CICR) is best known from cardiac muscle, but it also occurs in neurons. Neuronal CICR uses both ryanodine receptors (RyR1, RyR2, RyR3 are all expressed in different neuronal populations) and IP3 receptors on the smooth endoplasmic reticulum.
- Amplification of small calcium signals – A small VGCC-mediated calcium influx can trigger RyR opening on the adjacent ER, releasing stored calcium and amplifying the signal. This is particularly important in dendrites and dendritic spines, where local calcium signals trigger plasticity-related gene expression.
- IP3-mediated release – Metabotropic glutamate receptors (mGluR1/5) and acetylcholine muscarinic receptors couple via Gq to phospholipase C, generating IP3 that opens IP3 receptors on the ER and releases stored calcium. This pathway contributes to slow synaptic responses and long-term plasticity.
- Long-term potentiation (LTP) – The canonical LTP mechanism at CA3-CA1 hippocampal synapses involves NMDA receptor activation, calcium entry through the NMDA channel pore, activation of CaMKII, and CaMKII-mediated insertion of AMPA receptors into the postsynaptic density. CICR amplification of the NMDA-mediated calcium signal is part of why CICR pharmacological blockade (e.g., with dantrolene) can interfere with memory consolidation in animal models.
- Calcium oscillations and gene expression – Some neurons exhibit calcium oscillations whose frequency encodes specific information to downstream signaling pathways. NFAT translocation to the nucleus, for example, is more sensitive to slow calcium oscillations, while CaMKIV is more sensitive to fast spikes. Different calcium signal "shapes" thus produce different transcriptional outputs.
The Neuromuscular Junction
The neuromuscular junction (NMJ) is the specialized cholinergic synapse between a motor neuron and a skeletal muscle fiber. It is arguably the best-studied synapse in biology and the prototype for understanding calcium-triggered transmitter release.
- Presynaptic structure – The motor nerve terminal contains active zones aligned over postsynaptic acetylcholine receptors. Each active zone has a row of P/Q-type calcium channels (CaV2.1 is the dominant subtype at adult mammalian NMJs) flanking dense bars of docked synaptic vesicles.
- Postsynaptic structure – The motor end plate has deep junctional folds that increase the surface area for acetylcholine receptor (AChR) packing. AChRs cluster at the crests of the folds. Voltage-gated sodium channels concentrate at the depths of the folds to ensure action potential propagation into the muscle fiber.
- Acetylcholine release per action potential – A single action potential releases approximately 50-100 vesicles ("quanta") of acetylcholine at the mammalian NMJ. Each vesicle contains roughly 10,000 acetylcholine molecules. The resulting end-plate potential is typically far above threshold (the "safety factor" of neuromuscular transmission), so every motor neuron action potential reliably produces a muscle action potential.
- Acetylcholinesterase – The synaptic cleft contains acetylcholinesterase (AChE), which hydrolyzes ACh within milliseconds, terminating the signal. AChE inhibitors (neostigmine, pyridostigmine) prolong ACh action and are used in myasthenia gravis and to reverse neuromuscular blockade after surgery.
- Myasthenia gravis – An autoimmune disease in which antibodies against the postsynaptic AChR (or against MuSK, the receptor's clustering partner) reduce functional AChR density. The safety factor is compromised, producing progressive weakness with sustained activity. Treatment includes pyridostigmine (AChE inhibitor), immunosuppression, and in selected cases thymectomy.
- Lambert-Eaton myasthenic syndrome (LEMS) – A paraneoplastic / autoimmune syndrome in which antibodies target presynaptic P/Q-type calcium channels, reducing calcium influx and thus acetylcholine release. Strength paradoxically improves with repeated activity (post-tetanic potentiation). Strongly associated with small cell lung cancer. Treated with 3,4-diaminopyridine (which prolongs the presynaptic action potential and increases calcium entry) plus immunosuppression and treatment of any underlying tumor.
Hypocalcemia — Chvostek, Trousseau, and Tetany
Acute hypocalcemia is one of medicine's most dramatic emergencies. The neurological manifestations arise because low extracellular ionized calcium destabilizes the resting membrane potential of nerve and muscle, making spontaneous action potentials more likely. The classical clinical signs are pathognomonic enough that calcium repletion can be initiated before the laboratory result returns:
- Perioral and acral paresthesias – Tingling around the mouth and in the fingertips and toes is often the first symptom, reflecting heightened excitability of sensory nerve fibers. Patients may describe a sensation of "pins and needles" or a "buzz" in these areas.
- Carpopedal spasm – Sustained, involuntary tonic contraction of the hand muscles produces the characteristic "obstetrician's hand" posture: wrist flexion, metacarpophalangeal joint flexion, interphalangeal joint extension, and adduction of the thumb. The feet may show similar fixed posture (plantar flexion, toe flexion).
- Trousseau's sign – Inflating a blood pressure cuff above systolic pressure for 3 minutes induces carpopedal spasm. The maneuver works by inducing ischemia of the median nerve trunk, which lowers its already-reduced firing threshold further. A positive Trousseau is highly specific for latent tetany.
- Chvostek's sign – Tapping over the facial nerve trunk 1-2 cm anterior to the earlobe produces ipsilateral facial muscle twitch. Sensitive but less specific than Trousseau (positive Chvostek occurs in 10-25% of normocalcemic adults).
- Laryngospasm and stridor – Sustained contraction of the laryngeal muscles can produce inspiratory stridor and, in severe cases, airway obstruction. This is one of the airway emergencies in severe acute hypocalcemia (e.g., after parathyroidectomy or in massive transfusion with citrate toxicity).
- Seizures – Hypocalcemia lowers seizure threshold; both partial and generalized tonic-clonic seizures can occur. The mechanism is the same destabilization of neuronal membranes that causes peripheral tetany, applied to the central nervous system.
- QT prolongation – Hypocalcemia prolongs the QT interval on ECG (specifically, the ST segment) and predisposes to torsades de pointes, particularly when combined with hypomagnesemia or QT-prolonging drugs.
- Causes – Post-parathyroidectomy hypoparathyroidism, vitamin D deficiency, chronic kidney disease, acute pancreatitis (saponification), hypomagnesemia (impairs PTH secretion and action), citrate toxicity from massive transfusion, sepsis-associated hypocalcemia, hungry bone syndrome after correcting chronic secondary hyperparathyroidism, and certain medications (bisphosphonates, calcitonin, denosumab, foscarnet, cisplatin).
- Treatment – Acute symptomatic hypocalcemia: IV calcium gluconate (preferred over calcium chloride for peripheral administration; calcium chloride contains more elemental calcium but is more sclerosing). Always assess and correct concurrent hypomagnesemia. Chronic hypocalcemia: oral calcium plus activated vitamin D (calcitriol or alfacalcidol); recombinant PTH (rhPTH 1-84) for selected hypoparathyroidism patients.
Calcium and Seizure Threshold
The relationship between extracellular calcium and neuronal excitability is one of the foundational principles of clinical neurophysiology. Extracellular Ca2+ binds to the outer surface of the neuronal membrane, screening negative charges on membrane phospholipids and effectively making the membrane potential "feel" more negative to voltage-sensing channels. When extracellular calcium falls:
- Voltage-gated sodium channels become more easily activated – Their voltage-dependence shifts in the hyperpolarizing direction, so a smaller depolarization is needed to open them. Spontaneous, ectopic action potential firing becomes more likely.
- Spike threshold falls – Across both peripheral and central neurons, the threshold for action potential generation drops, predisposing to hyperexcitability syndromes ranging from peripheral tetany to generalized seizures.
- Seizure threshold drops – In otherwise normal brains, hypocalcemia below approximately 7 mg/dL (1.75 mmol/L) total calcium (or correspondingly low ionized calcium) markedly increases seizure risk. Below 6 mg/dL, generalized tonic-clonic seizures are common.
- EEG patterns in hypocalcemia – Diffuse slowing, generalized spike-wave discharges, and frank seizure activity correlate with the depth of hypocalcemia. EEG findings reverse with calcium correction.
Conversely, hypercalcemia (calcium >14 mg/dL or so) tends to produce CNS depression, lethargy, and coma rather than seizures, because the increased extracellular calcium screens the membrane more effectively and raises spike threshold.
The clinical implication is that any patient presenting with new-onset seizures should have serum calcium (and magnesium) checked as part of the basic metabolic workup. Hypocalcemic seizures resolve with calcium repletion and do not require chronic antiepileptic drug therapy.
Calcium Channelopathies (LEMS, Hemiplegic Migraine, CPVT)
The voltage-gated calcium channel genes have produced a rich and growing list of disease associations, both autoimmune and inherited. These channelopathies illuminate the specific roles of individual channel subtypes in normal neural function.
- Lambert-Eaton myasthenic syndrome (LEMS) – Autoantibodies against presynaptic P/Q-type CaV2.1 channels at the neuromuscular junction reduce ACh release. Two-thirds of cases are paraneoplastic (most commonly small cell lung cancer). Treatment includes treating the underlying tumor, 3,4-diaminopyridine (amifampridine) to prolong the presynaptic action potential, and immunotherapy.
- Familial hemiplegic migraine type 1 (FHM1) – Gain-of-function mutations in CACNA1A (CaV2.1) cause migraine with aura accompanied by reversible hemiparesis. Cortical spreading depression is more easily triggered in these patients. Notably, FHM1 mutations also cause episodic ataxia type 2 and spinocerebellar ataxia type 6.
- Catecholaminergic polymorphic ventricular tachycardia (CPVT) – Gain-of-function mutations in RyR2 (cardiac ryanodine receptor) cause diastolic SR calcium leak, delayed afterdepolarizations, and stress-triggered ventricular arrhythmia. Although primarily a cardiac disease, the mechanism is informative for understanding RyR-dependent neural calcium signaling.
- Timothy syndrome – Gain-of-function mutations in CACNA1C (CaV1.2) cause QT prolongation, syndactyly, autism spectrum disorder, and developmental delay. Demonstrates the breadth of L-type calcium channel function across cardiac, neural, and developmental contexts.
- Episodic ataxia type 2 – Loss-of-function CACNA1A mutations cause attacks of cerebellar ataxia, often responsive to acetazolamide. The same gene is involved in both gain-of-function (FHM1) and loss-of-function (EA2) syndromes depending on the specific mutation.
- Absence epilepsy and T-type channels – T-type calcium channels (CACNA1G, CACNA1H, CACNA1I encoding CaV3.1, 3.2, 3.3) contribute to the thalamocortical oscillations underlying absence seizures. Ethosuximide, the drug of choice for absence epilepsy, blocks T-type channels.
Calcium Signaling in Aging and Cognition
The "calcium hypothesis of aging" proposes that progressive dysregulation of intracellular calcium signaling contributes to age-related neurological decline. Multiple lines of evidence support this:
- Calcium load and Parkinson's disease – Substantia nigra dopamine neurons use CaV1.3 L-type channels for pacemaker activity, producing a chronic calcium load that may contribute to their selective vulnerability in Parkinson's. The STEADY-PD trial tested whether isradipine (an L-type calcium channel blocker) could slow Parkinson's progression; the trial was negative for the primary endpoint, though mechanistic interest in calcium handling persists.
- Hippocampal LTP changes with age – Long-term potentiation, the cellular correlate of memory, becomes less robust in aged animals. Part of this reflects shifts in calcium handling: increased L-type calcium channel density in aging neurons, altered CICR amplitude, and changes in CaMKII responsiveness.
- Alzheimer's disease and calcium – Mutations in presenilin (gamma-secretase complex) that cause familial Alzheimer's disease also disrupt ER calcium handling. Amyloid-beta peptides can permeabilize membranes to calcium. The "calcium hypothesis of Alzheimer's" is one of several active research threads complementing the amyloid and tau-centered models.
- Calcium-binding proteins and neuronal vulnerability – Calretinin-positive and parvalbumin-positive interneurons (which express high levels of calcium-buffering proteins) are relatively spared in some neurodegenerative diseases, suggesting that calcium buffer capacity is protective.
Key Research Papers
- Sudhof TC (2012). Calcium control of neurotransmitter release. Cold Spring Harbor Perspectives in Biology. — DOI: 10.1101/cshperspect.a011353
- Katz B, Miledi R (1967). The timing of calcium action during neuromuscular transmission. Journal of Physiology. — PubMed
- Catterall WA (2011). Voltage-gated calcium channels. Cold Spring Harbor Perspectives in Biology. — DOI: 10.1101/cshperspect.a003947
- Berridge MJ (1998). Neuronal calcium signaling. Neuron. — DOI: 10.1016/S0896-6273(00)80510-3
- Augustine GJ (2001). How does calcium trigger neurotransmitter release? Current Opinion in Neurobiology. — DOI: 10.1016/S0959-4388(00)00219-6
- Brose N, Petrenko AG, Sudhof TC, Jahn R (1992). Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. — DOI: 10.1126/science.1589771
- Tarr TB, Dittrich M, Meriney SD (2013). Are unreliable release mechanisms conserved from NMJ to CNS? Trends in Neurosciences. — DOI: 10.1016/j.tins.2012.09.009
- Cooper EC, Jan LY (1999). Ion channel genes and human neurological disease: recent progress, prospects, and challenges. PNAS. — DOI: 10.1073/pnas.96.9.4759
- Vincent A, Lang B, Newsom-Davis J (1989). Autoimmunity to the voltage-gated calcium channel underlies the Lambert-Eaton myasthenic syndrome. Trends in Neurosciences. — DOI: 10.1016/0166-2236(89)90123-3
- Ophoff RA, Terwindt GM, Vergouwe MN, et al. (1996). Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. — DOI: 10.1016/S0092-8674(00)81373-2
- Cooper MS, Gittoes NJ (2008). Diagnosis and management of hypocalcaemia. BMJ. — DOI: 10.1136/bmj.39582.589433.BE
- Khosla S (2008). Hypercalcemia and hypocalcemia. In: Harrison's Principles of Internal Medicine. — PubMed search
- Surmeier DJ, Schumacker PT (2013). Calcium, bioenergetics, and neuronal vulnerability in Parkinson's disease. Journal of Biological Chemistry. — DOI: 10.1074/jbc.R112.410530
PubMed Topic Searches
- PubMed: Synaptotagmin / SNARE
- PubMed: VGCC presynaptic release
- PubMed: LEMS / P/Q channel autoimmunity
- PubMed: Familial hemiplegic migraine
- PubMed: Hypocalcemia / tetany / seizure
- PubMed: Calcium hypothesis of aging
External Authoritative Resources
- Neuroscience (Purves), 2nd ed. — Neurotransmitter Release (NCBI Bookshelf)
- NIH Office of Dietary Supplements — Calcium Fact Sheet
- Linus Pauling Institute — Calcium
Connections
- Calcium (Main Page)
- Calcium Benefits Hub
- Calcium for Bone Health
- Calcium for Cardiovascular Health
- Calcium for Muscle Function
- Magnesium (PTH cofactor)
- Vitamin D3
- Vitamin K2
- Hyperparathyroidism
- Epilepsy / Seizure Disorders
- Parkinson's Disease
- Alzheimer's Disease
- Myasthenia Gravis
- Migraine
- Stroke / Excitotoxicity
- Cramp Prevention
- All Minerals