Interstitial Lung Disease: History and Discovery
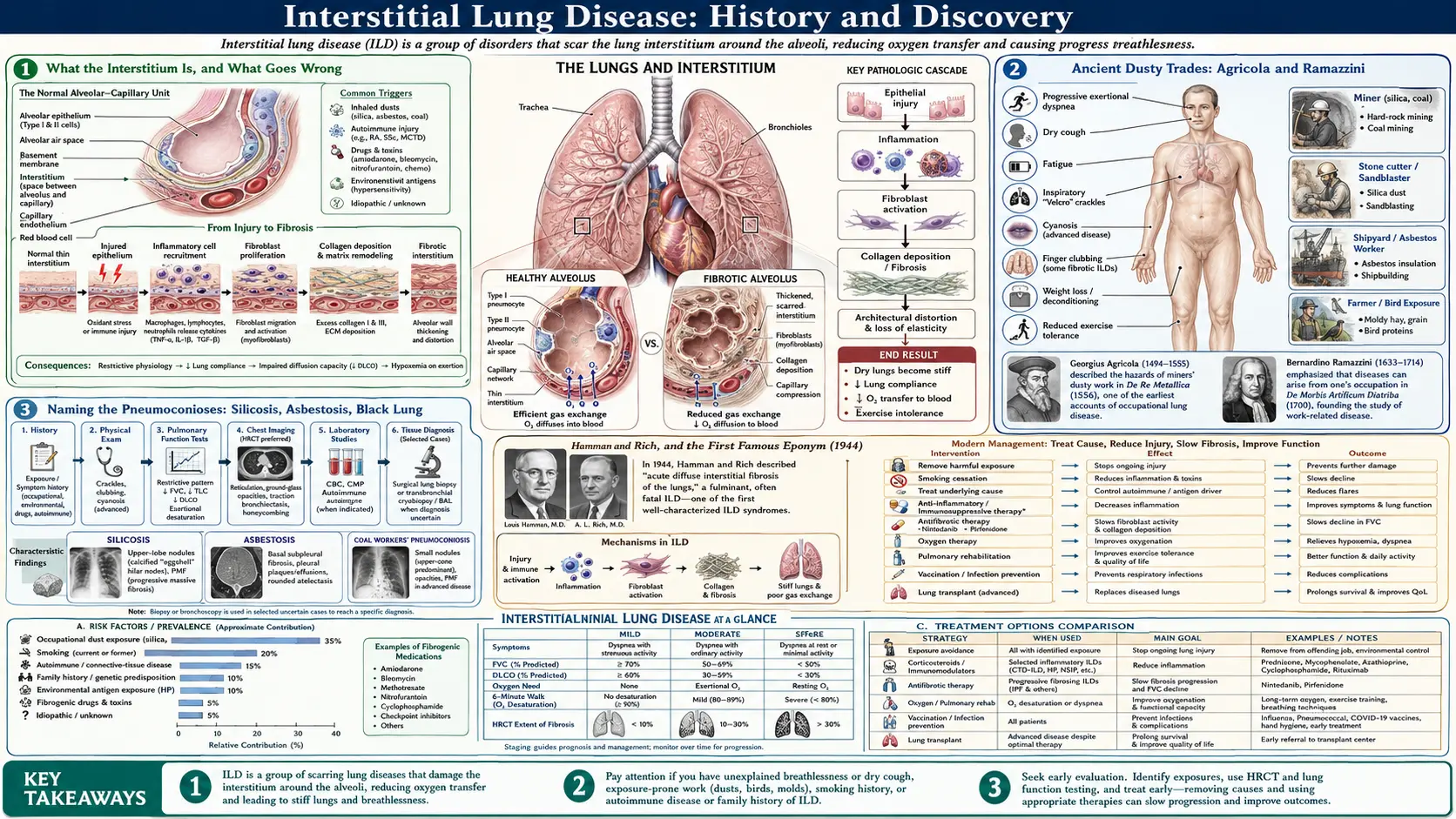
Interstitial lung disease (ILD) is not one illness but a large family of more than two hundred disorders that inflame and scar the interstitium — the delicate scaffolding of tissue that surrounds the lung's air sacs (alveoli) and carries its blood vessels. When this tissue thickens, stiffens, and scars, the lung loses its softness, breathing becomes hard work, and oxygen struggles to cross into the blood; the end result is often pulmonary fibrosis. The history of ILD is therefore really several histories braided together: the ancient story of the dusty trades and their pneumoconioses, the twentieth-century effort to recognize and name fibrosis "of unknown cause," the pathologists' long labor to classify the bewildering patterns seen down the microscope, and the very recent arrival of drugs that can finally slow the scarring. This page traces those threads honestly, naming a discoverer or a date only where the record supports it.
Table of Contents
- What the Interstitium Is, and What Goes Wrong
- Ancient Dusty Trades: Agricola and Ramazzini
- Naming the Pneumoconioses: Silicosis, Asbestosis, Black Lung
- Hamman and Rich, and the First Famous Eponym (1944)
- Liebow, Carrington, and the Birth of Classification (1969)
- Idiopathic Pulmonary Fibrosis and the UIP Pattern
- Related Family Members: Hypersensitivity Pneumonitis and Sarcoidosis
- High-Resolution CT and the Modern Classifications
- The Antifibrotic Era: Pirfenidone and Nintedanib (2014)
- Research Papers and References
- Connections
- Featured Videos
What the Interstitium Is, and What Goes Wrong
To follow the history it helps to picture the tissue at the center of it. A healthy lung is mostly air and a paper-thin membrane: roughly three hundred million alveoli, each wrapped in capillaries, with only a microscopically thin sheet of tissue separating inhaled air from flowing blood. That separating sheet — together with the supporting framework that holds the air sacs open — is the interstitium. In a normal lung it is so thin that oxygen and carbon dioxide cross it almost instantly. The whole point of the lung's architecture is to keep this barrier as thin and as vast as possible.
Interstitial lung disease is what happens when that barrier is injured and the body's repair response goes wrong. Instead of healing cleanly, the tissue fills with inflammatory cells and then with scar — tough, fibrous collagen laid down where airy membrane used to be. The lung becomes stiff and small (doctors call this a restrictive pattern, the opposite of the obstructive blockage seen in asthma or COPD), and the thickened barrier slows the passage of oxygen. The result is the classic picture: relentless breathlessness on exertion, a dry persistent cough, fatigue, and on examination a fine crackling sound at the lung bases that clinicians liken to the pulling-apart of Velcro.
The reason ILD has such a tangled history is that this same end-point — scarred interstitium — can be reached by dozens of different roads. Inhaled mineral dust, mouldy hay, an autoimmune disease, a drug reaction, radiation, or no identifiable cause at all can each end in fibrosis that looks broadly similar to the patient and sometimes even to the pathologist. Untangling those roads, giving them names, and learning which ones could be prevented or treated is the work that the rest of this page describes.
Ancient Dusty Trades: Agricola and Ramazzini
The oldest chapter of the ILD story belongs to the working lung. Long before anyone could see scar tissue under a microscope, people noticed that certain trades stole the breath of those who practiced them — miners, stonecutters, potters, and millers grew short of breath and died young. The earliest sustained written observation comes from the German scholar Georgius Agricola (Georg Bauer), whose great mining treatise De Re Metallica was published in 1556. Agricola described the lung complaints of miners in the Carpathian mountains and grasped the essential link — that dust stirred up by digging entered the lungs and caused shortness of breath. It is among the earliest, and arguably the first clear, descriptions of a dust-induced lung disease.
The next landmark is the Italian physician Bernardino Ramazzini, often called the father of occupational medicine. His De Morbis Artificum Diatriba ("Diseases of Workers"), published in 1700, was the first systematic survey of how specific occupations harmed the body. Ramazzini catalogued the dusty trades — stonemasons, potters, miners — and described the cough and breathlessness of workers who inhaled mineral and vegetable dusts, urging physicians to ask every patient, "What is your trade?" His work turned scattered folk knowledge into a method.
It is worth being precise about what these men did and did not establish. Agricola and Ramazzini observed and described the diseases of dusty work centuries before the cause was understood at the level of tissue and cell; they did not name "silicosis" or know that crystalline silica was the culprit. Their lasting contribution was the insight that the air of certain workplaces was itself dangerous to the lung — the founding observation of every occupational interstitial disease that followed.
Naming the Pneumoconioses: Silicosis, Asbestosis, Black Lung
The dust diseases as a group are called the pneumoconioses (from the Greek for "dust in the lungs"). Silicosis — fibrosis from inhaling crystalline silica in stone, sand, and ore — is the archetype, observed in substance since Agricola but named only much later. The term "silicosis" is generally credited to the Italian pathologist Achille Visconti, who used it around 1870 after examining the badly fibrosed lungs of a stonecutter; the word fixed in medical language the connection between silica dust and lung scarring that the old miners' complaints had only hinted at.
Asbestosis — fibrosis caused by asbestos fibres — is a twentieth-century discovery. A landmark moment came in 1924, when the British physician W. E. Cooke reported the death of a textile worker, Nellie Kershaw, from lung fibrosis he attributed to inhaled asbestos dust; in a follow-up paper in 1927 he proposed the name "pulmonary asbestosis" for the condition. Cooke's reports are widely regarded as the first clear scientific description of the disease and the origin of its name, and they helped drive the earliest industrial dust regulations.
The third great occupational fibrosis is coal workers' pneumoconiosis, the disease behind the grim popular name "black lung." The blackening and scarring of coal miners' lungs had been noted by pathologists in the nineteenth century, but its recognition as a distinct, compensable occupational disease — separate from silicosis and from tuberculosis — was hard-won and came largely in the twentieth century through the campaigning of miners and mining communities. Together silicosis, asbestosis, and coal workers' pneumoconiosis form the occupational branch of the ILD family: diseases whose cause was, from the start, no mystery at all, and whose history is as much about industry, labour, and law as about medicine.
Hamman and Rich, and the First Famous Eponym (1944)
While the dust diseases had obvious causes, physicians kept encountering patients whose lungs scarred for no apparent reason. The most influential early account of such a case came from two physicians at the Johns Hopkins Hospital in Baltimore: the clinician Louis Hamman and the pathologist Arnold Rich. They first reported the condition in 1935 and described it more fully in a paper titled "Acute diffuse interstitial fibrosis of the lungs," published in the Bulletin of the Johns Hopkins Hospital in 1944. Their report centered on a small series of patients who had died swiftly — over weeks — of a fulminant illness whose lungs at autopsy showed a striking overgrowth of interstitial fibrous tissue.
The disease became widely known by the eponym Hamman–Rich syndrome. For decades the name was used loosely, sometimes for any rapidly progressive idiopathic pulmonary fibrosis, which caused real confusion in the literature. Modern pathology has resolved this: what Hamman and Rich actually described — an acute, rapidly fatal interstitial process — is now recognized as a specific entity called acute interstitial pneumonia (AIP), characterized microscopically by diffuse alveolar damage and clinically resembling an idiopathic form of acute respiratory distress syndrome. It is distinct from the chronic, slowly progressive idiopathic pulmonary fibrosis with which the eponym was once carelessly merged.
The historical importance of the Hamman–Rich report is less the specific disease than the precedent. It was an early, careful clinical-and-pathological description of interstitial fibrosis arising without dust, infection, or other obvious cause — an "idiopathic" interstitial disease — and it put such cases firmly on the medical map. Almost everything that followed in the study of idiopathic interstitial pneumonias can be read as an effort to do more precisely what Hamman and Rich first attempted: to separate one pattern of unexplained lung scarring from another.
Liebow, Carrington, and the Birth of Classification (1969)
If the pneumoconioses gave ILD its causes and Hamman–Rich gave it a famous case, it was the pathologist Averill A. Liebow who gave it an order. Working from the microscope, Liebow recognized that the "idiopathic interstitial pneumonias" were not a single disease but several, each with a characteristic tissue pattern, a different clinical course, and a different prognosis. In 1969, Liebow and his colleague Charles B. Carrington published the first influential histological classification of these disorders — the framework on which all later schemes were built.
Liebow's original classification sorted the idiopathic interstitial pneumonias into a handful of patterns identified by acronym, the most important of which was usual interstitial pneumonia (UIP) — a term Liebow himself coined in the 1960s for the commonest pattern of fibrotic scarring, with its patchy, "temporally heterogeneous" mix of old scar, active fibrosis, and honeycombed lung. His scheme also named desquamative interstitial pneumonia (DIP), lymphoid (lymphocytic) interstitial pneumonia (LIP), and other patterns. The central idea — that the precise look of the tissue predicts how the patient will fare — was a genuine conceptual advance.
Liebow's categories have been debated, renamed, and refined ever since; some of his original entities have been dropped or reassigned as understanding improved. But the principle he established — that interstitial lung diseases must be distinguished by reproducible morphological pattern, and that pattern carries prognosis — is the foundation of every modern classification, and the term UIP he introduced remains central to the diagnosis of pulmonary fibrosis today.
Idiopathic Pulmonary Fibrosis and the UIP Pattern
Of all the interstitial lung diseases, the one that has come to dominate research and to define the field is idiopathic pulmonary fibrosis (IPF) — chronic, progressive scarring of the lung with no identifiable cause. Carving IPF out as a specific diagnosis, rather than a vague label for any unexplained fibrosis, was one of the central achievements of twentieth-century pulmonology, and it was accomplished largely by tying the clinical disease to Liebow's UIP tissue pattern. In current usage, IPF is essentially defined as the clinical illness that arises from a UIP pattern in the lung when every other cause has been excluded.
The naming of the disease itself has a history. In Britain it was for many years called cryptogenic fibrosing alveolitis (CFA), a term that emphasized inflammation of the alveoli "of hidden cause"; in North America the term "idiopathic pulmonary fibrosis" prevailed. As research showed that the disease was driven more by aberrant scarring than by primary inflammation, the inflammation-centered name fell out of favour, and international agreement eventually settled on IPF defined by the UIP pattern — a shift in name that mirrored a real shift in understanding of the disease's biology.
That shift mattered enormously for patients, because it changed how the disease was treated. For decades, on the assumption that fibrosis was the end-stage of inflammation, IPF was treated with corticosteroids and other immunosuppressants — an approach that was later shown, in carefully controlled trials, to be ineffective and at times harmful for true IPF. Recognizing IPF as a fundamentally fibrotic rather than inflammatory disease cleared the way for an entirely different therapeutic strategy: drugs aimed directly at the scarring process, which finally arrived in 2014 and are described below.
Related Family Members: Hypersensitivity Pneumonitis and Sarcoidosis
The ILD family includes major members whose interstitial scarring is driven not by dust or by chance but by the immune system. The clearest example is hypersensitivity pneumonitis (once called extrinsic allergic alveolitis) — an allergic-immune reaction in the lung to repeatedly inhaled organic antigens such as mould spores, bird proteins, or bacteria growing in damp vegetable matter. Its classic form, farmer's lung, was described in detail by the British physician Munro Campbell in 1932, who linked the breathlessness and fever of Westmorland farmers to their handling of mouldy hay. Hypersensitivity pneumonitis showed, vividly, that an everyday environmental exposure could trigger interstitial inflammation and, with time, fibrosis — and that removing the exposure could halt or reverse it.
A second great immune-related member is sarcoidosis, a disease in which clusters of inflammatory cells called granulomas form in the lungs and other organs. Its history reaches back to the English surgeon Jonathan Hutchinson, who in 1877 described the skin lesions of a patient in a case that is generally credited as the first report, and to the Norwegian dermatologist Caesar Boeck, who in 1899 coined the term "sarkoid" because the skin nodules resembled sarcoma yet were benign. Only later was it understood that this was a single multi-organ disease with a strong predilection for the lung and its lymph nodes. Because sarcoidosis is covered in depth in its own article, it is mentioned here chiefly to place it within the interstitial family.
Beyond these, the interstitial family extends to the lung disease of the autoimmune connective-tissue disorders — rheumatoid arthritis, systemic sclerosis (scleroderma), lupus, and related conditions can all scar the interstitium — and to fibrosis triggered by certain drugs and by radiation. The recognition that so many different diseases share a final common pathway of interstitial scarring is precisely what makes "interstitial lung disease" a useful umbrella, and what has driven the search for treatments that target the scarring itself rather than only its many separate causes.
High-Resolution CT and the Modern Classifications
For most of the twentieth century, distinguishing one interstitial disease from another required either a chest X-ray too crude to show fine detail or a surgical lung biopsy too invasive to perform freely. The transformative technology was high-resolution computed tomography (HRCT), which from the 1980s onward let physicians see the texture of the lung in exquisite detail without surgery — the reticular lines, ground-glass haze, traction bronchiectasis, and the characteristic clustered cysts of "honeycombing." HRCT made it possible to recognize the UIP pattern non-invasively, and it revolutionized the diagnosis of ILD, often replacing the need for biopsy entirely.
With better imaging came better consensus. In 2002, the American Thoracic Society and the European Respiratory Society (ATS/ERS) published a landmark multidisciplinary classification of the idiopathic interstitial pneumonias, defining a standard set of entities — including IPF (UIP pattern), nonspecific interstitial pneumonia (NSIP), acute interstitial pneumonia (the Hamman–Rich entity), desquamative interstitial pneumonia, cryptogenic organizing pneumonia, and others — with agreed names and diagnostic criteria. The scheme insisted that diagnosis should be made not by any single specialist but by clinicians, radiologists, and pathologists working together: the multidisciplinary diagnosis that remains the gold standard.
The classification was updated in 2013, refining the categories, formally recognizing additional patterns, and adding a new entity (pleuroparenchymal fibroelastosis). The deeper change embodied in these statements was philosophical: ILD came to be understood not as a fixed list of diseases but as a spectrum of behaviours, best captured by combining the patient's history and exposures, the HRCT appearance, and where necessary the biopsy. This integrated, imaging-led approach is the framework within which interstitial lung disease is diagnosed today.
The Antifibrotic Era: Pirfenidone and Nintedanib (2014)
For almost the entire history recounted above, medicine could describe and classify pulmonary fibrosis far better than it could treat it. For IPF in particular there was, in honest terms, no drug that slowed the disease; care was supportive — oxygen, pulmonary rehabilitation, treatment of complications, and for suitable patients lung transplantation. The old standby of corticosteroids and immunosuppression, once given almost reflexively, was shown by controlled trials to be useless or harmful in genuine IPF. The central need was a medicine that could act on the scarring itself.
That need was met in 2014, a watershed year, when the U.S. Food and Drug Administration approved two antifibrotic drugs for IPF on the same day, 15 October 2014: pirfenidone (marketed as Esbriet) and nintedanib (marketed as Ofev). Pirfenidone is an oral agent with anti-fibrotic and anti-inflammatory actions; nintedanib is a tyrosine-kinase inhibitor that blocks growth-factor signalling pathways that drive fibrosis. Neither cures the disease or reverses existing scar, but each was shown in randomized trials to slow the decline in lung function — roughly halving the rate at which forced vital capacity fell — which for a relentlessly progressive disease was a genuine and long-awaited advance.
The significance of 2014 is hard to overstate for patients with IPF: it marked the transition from a disease that could only be managed to one that could, at last, be actively treated. In the years since, nintedanib in particular has been shown to slow other progressive fibrosing interstitial lung diseases beyond classic IPF, broadening the antifibrotic strategy across the wider ILD family. The arc of this history — from Agricola's breathless miners, through the naming of the dust diseases, the careful eponyms and classifications of the twentieth century, and the imaging revolution, to the first drugs that touch the scar — is a reminder of how long it can take to move from recognizing a disease to being able to do something about it, and of how much the effort is worth.
Research Papers and References
The list below combines key peer-reviewed historical and clinical reviews with curated PubMed topic-search links into the literature on the history of interstitial lung disease, pulmonary fibrosis, the pneumoconioses, and antifibrotic therapy. Historical primary texts — Agricola's De Re Metallica (1556) and Ramazzini's De Morbis Artificum Diatriba (1700) — are named in the article as historical sources rather than as modern citations. Each link opens in a new tab.
- Mueller-Mang C, Grosse C, Schmid K, Stiebellehner L, Bankier AA. What every radiologist should know about idiopathic interstitial pneumonias. RadioGraphics. 2007;27(3):595-615. — doi:10.1148/rg.273065130
- American Thoracic Society / European Respiratory Society. International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med. 2002;165(2):277-304. — doi:10.1164/ajrccm.165.2.ats01
- Travis WD, Costabel U, Hansell DM, et al. An Official ATS/ERS Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med. 2013;188(6):733-748. — doi:10.1164/rccm.201308-1483ST
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al. (ASCEND Study Group). A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N Engl J Med. 2014;370(22):2083-2092. — doi:10.1056/NEJMoa1402582
- Richeldi L, du Bois RM, Raghu G, et al. (INPULSIS Trial Investigators). Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N Engl J Med. 2014;370(22):2071-2082. — doi:10.1056/NEJMoa1402584
- Mason RJ, Schwarz MI, Hunninghake GW, Musson RA. Pharmacological therapy for idiopathic pulmonary fibrosis — past, present, and future. — PubMed: Hamman-Rich syndrome / acute interstitial pneumonia history
- Liebow and Carrington classification of the interstitial pneumonias (historical perspective) — PubMed: Liebow-Carrington classification / UIP
- History and naming of silicosis as an occupational lung disease — PubMed: silicosis history (Agricola, Ramazzini)
- Asbestosis — Cooke and the early description of asbestos-induced pulmonary fibrosis — PubMed: asbestosis history (Cooke)
- Coal workers' pneumoconiosis ("black lung") — history and recognition — PubMed: coal workers' pneumoconiosis history
- Farmer's lung and hypersensitivity pneumonitis — Campbell 1932 and the history of the disease — PubMed: farmer's lung / hypersensitivity pneumonitis history
- High-resolution CT in the diagnosis of interstitial lung disease and the UIP pattern — PubMed: HRCT in ILD diagnosis
- Idiopathic pulmonary fibrosis / cryptogenic fibrosing alveolitis — terminology and concept — PubMed: IPF / cryptogenic fibrosing alveolitis
- Antifibrotic therapy in progressive fibrosing interstitial lung disease (pirfenidone, nintedanib) — PubMed: antifibrotic therapy in ILD
External Authoritative Resources
- NHLBI (National Heart, Lung, and Blood Institute) — Interstitial Lung Diseases
- MedlinePlus — Interstitial Lung Diseases
- PubMed — History of interstitial lung disease and pulmonary fibrosis
Connections
- Pulmonology
- Interstitial Lung Disease (main article)
- All Conditions
- Sarcoidosis
- Pulmonary Hypertension
- COPD
- Pulmonary Embolism