Cystic Fibrosis
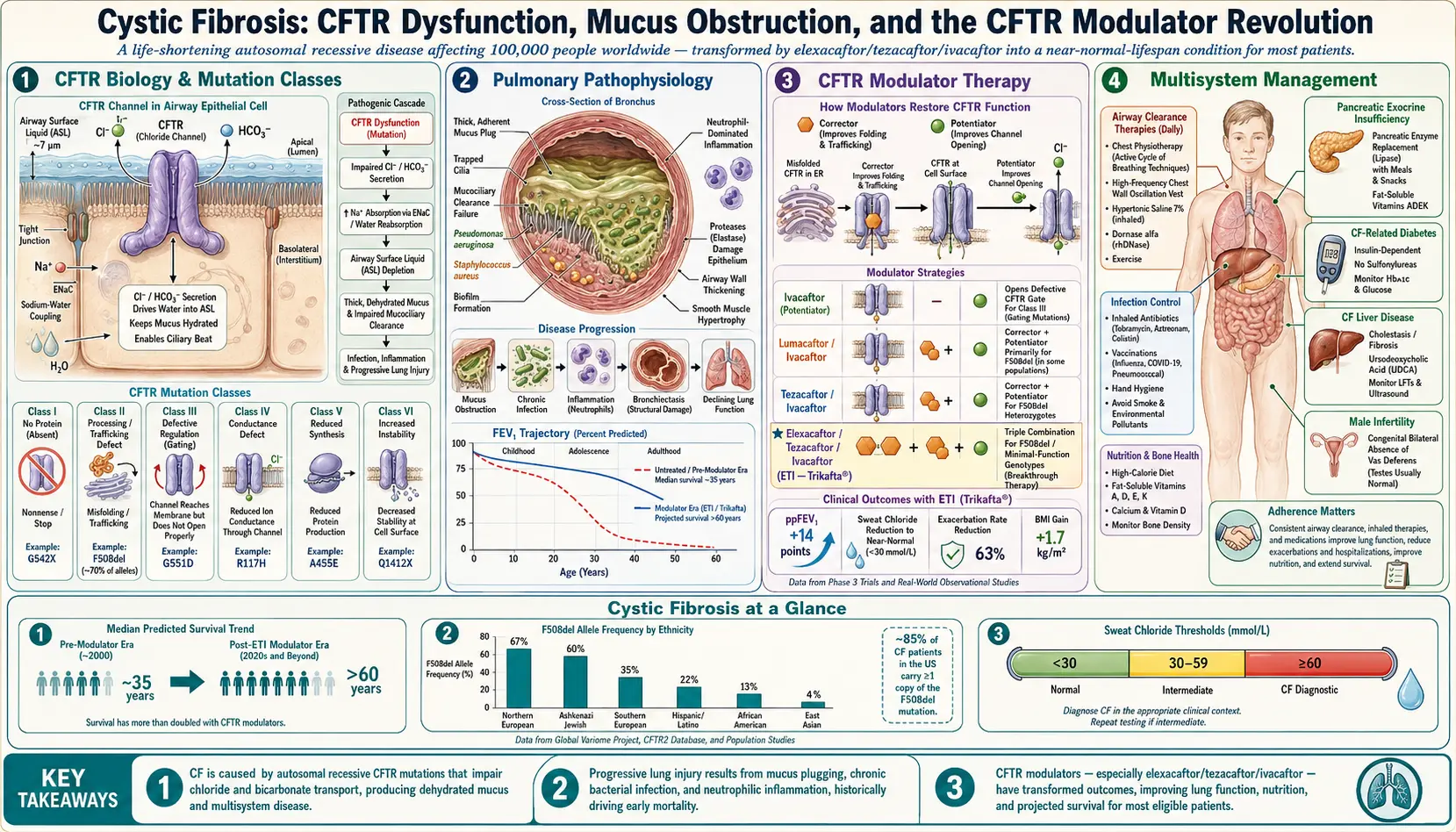
Table of Contents
- What is Cystic Fibrosis?
- Causes of Cystic Fibrosis
- Common Symptoms of Cystic Fibrosis
- Complications of Cystic Fibrosis
- Diagnosis
- Treatment Options
- Prevention and Management Strategies
- Living with Cystic Fibrosis
- Research Papers
- Connections
- Featured Videos
What is Cystic Fibrosis?
Cystic fibrosis (CF) is a genetic disorder that primarily affects the lungs and digestive system. It is caused by mutations in the CFTR (cystic fibrosis transmembrane conductance regulator) gene, leading to the production of thick, sticky mucus that can clog the airways and trap bacteria, resulting in repeated lung infections and breathing difficulties. CF can also affect the pancreas, liver, and other organs, leading to a variety of health complications.
Causes of Cystic Fibrosis
- Genetic inheritance: CF is an autosomal recessive disorder, which means a person must inherit two defective CFTR genes (one from each parent) to develop the disease.
- CFTR gene mutation: This mutation disrupts the function of the CFTR protein, affecting the transport of salt and water in and out of cells, resulting in thick mucus formation.
Common Symptoms of Cystic Fibrosis
- Persistent coughing with thick mucus.
- Frequent lung infections, such as pneumonia or bronchitis.
- Wheezing or shortness of breath
- Salty-tasting skin
- Poor growth and weight gain despite adequate food intake.
- Digestive issues: Including greasy, bulky stools and constipation.
- Sinus infections and nasal polyps.
Complications of Cystic Fibrosis
- Chronic lung infections: Due to mucus buildup that traps bacteria.
- Bronchiectasis: Damage to the airways that makes it difficult to clear mucus.
- Pancreatic damage: Thick mucus can block ducts in the pancreas, affecting enzyme release and nutrient absorption.
- Diabetes: CF-related diabetes can develop due to damage to the pancreas.
- Liver disease: Blocked bile ducts can lead to liver problems.
- Infertility in males: Due to the absence or blockage of the vas deferens.
Diagnosis
- Newborn screening: CF is often diagnosed shortly after birth through blood tests.
- Sweat test: Measures the amount of salt in sweat; high salt levels indicate CF.
- Genetic testing: Identifies mutations in the CFTR gene.
- Chest X-rays and CT scans: To assess lung damage and detect complications.
- Lung function tests: Evaluate how well the lungs are working.
Treatment Options
- Airway clearance techniques: Includes chest physiotherapy and devices that help remove mucus from the lungs.
- Medications:
- Bronchodilators: Open the airways to improve breathing.
- Mucus-thinning drugs: Help thin mucus so it can be coughed out more easily.
- Antibiotics: Used to treat and prevent lung infections.
- CFTR modulators: Such as ivacaftor and lumacaftor to improve the function of the defective CFTR protein.
- Enzyme supplements: Aid in digestion and nutrient absorption.
- High-calorie diet: Necessary to maintain energy levels and support growth.
- Lung transplant: Considered for severe cases when lung function is significantly compromised.
Prevention and Management Strategies
- Regular follow-ups: Frequent check-ups to monitor and manage symptoms and complications.
- Good nutrition: High-calorie, nutrient-dense foods to support growth and energy needs.
- Exercise: Regular physical activity to improve lung function.
- Hydration: Helps keep mucus thin and easier to expel.
- Avoid smoke and pollutants: To reduce the risk of lung infections and irritation.
Living with Cystic Fibrosis
Advancements in treatment have significantly improved the quality of life and life expectancy for people with cystic fibrosis. Early diagnosis, comprehensive treatment plans, and supportive care can help manage symptoms, reduce complications, and enable individuals to live fulfilling lives.
References & Research
Historical Background
Cystic fibrosis was first recognized as a distinct disease by Dorothy Hansine Andersen in 1938, who described its pathology in a landmark paper connecting pancreatic lesions with lung disease in children. The underlying genetic defect in the CFTR gene was identified in 1989 by Lap-Chee Tsui, John Riordan, and Francis Collins, opening the door to targeted molecular therapies.
Key Research Papers
- Elborn JS. Cystic fibrosis. Lancet. 2016;388(10059):2519-2531.
- Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066-1073.
- Middleton PG, Mall MA, Dřícek P, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele (TRIKAFTA). N Engl J Med. 2019;381(19):1809-1819.
- Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation (STRIVE). N Engl J Med. 2011;365(18):1663-1672.
- Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR (TRAFFIC and TRANSPORT). N Engl J Med. 2015;373(3):220-231.
- Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2013;187(7):680-689.
- Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis (Pulmozyme). N Engl J Med. 1994;331(10):637-642.
- Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa. JAMA. 2003;290(13):1749-1756.
- Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation. Lancet. 2019;394(10212):1940-1948.
- Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del (EVOLVE). N Engl J Med. 2017;377(21):2013-2023.
- Cystic Fibrosis Foundation Patient Registry. 2020 Annual Data Report. Bethesda, MD: Cystic Fibrosis Foundation. 2021.
- Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16(1):45-56.
Research Papers
The following PubMed topic searches surface the current peer-reviewed literature on Cystic Fibrosis. Each link opens a live PubMed query; results update as new papers are indexed.
- PubMed search: cystic fibrosis CFTR
- PubMed search: cystic fibrosis modulator therapy
- PubMed search: ivacaftor tezacaftor elexacaftor
- PubMed search: cystic fibrosis lung transplantation
- PubMed search: cystic fibrosis pancreatic insufficiency
- PubMed search: cystic fibrosis sweat chloride test
- PubMed search: cystic fibrosis newborn screening
- PubMed search: cystic fibrosis pseudomonas aeruginosa
- PubMed search: cystic fibrosis nutritional management
- PubMed search: cystic fibrosis related diabetes
- PubMed search: cystic fibrosis airway clearance
- PubMed search: cystic fibrosis gene therapy
Connections
- Pulmonology
- CRISPR: How Gene Editing Works — interactive animation
- Inheritance: Dominant, Recessive and Punnett Squares — interactive animation
- Your Airway’s Self-Cleaning Escalator (& the Cough) — interactive animation
- Pneumonia
- Diabetes
- COPD
- Vitamin D3
- Vitamin E
- Vitamin A
- Vitamin K
- Pseudomonas Aeruginosa
- Liver Disease
- Shortness of Breath
- Pancreatitis
- Infertility
- Interstitial Lung Disease
- Pulmonary Hypertension
- Inflammatory Markers
- Cirrhosis
- Constipation
- Chronic Cough