Polycythemia Vera
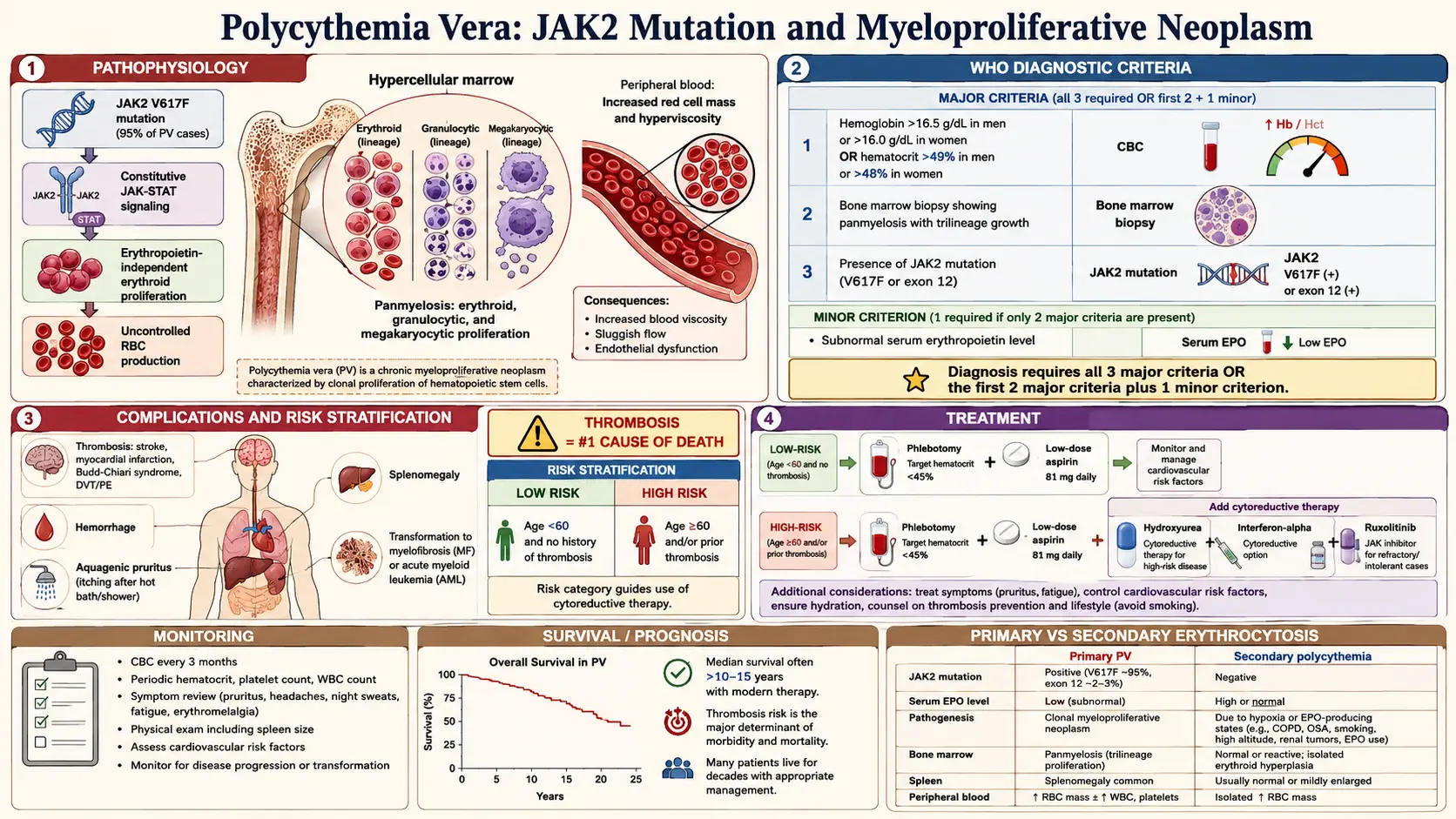
Table of Contents
- Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research and Advances
- Research Papers
- Connections
- Featured Videos
1. Overview
Polycythemia vera (PV) is a chronic myeloproliferative neoplasm (MPN) characterized by clonal expansion of a multipotent hematopoietic stem cell, resulting in autonomous, erythropoietin-independent overproduction of red blood cells (erythrocytosis) and, to a variable degree, excess white blood cells and platelets. It was first described by Louis Henri Vaquez in 1892 and later systematically characterized by William Osler. PV is classified by the World Health Organization (WHO) among the Philadelphia chromosome-negative myeloproliferative neoplasms alongside essential thrombocythemia (ET) and primary myelofibrosis (PMF).
The hallmark molecular lesion — a point mutation in the JAK2 gene (predominantly JAK2 V617F) — is present in virtually all cases and is central to both pathogenesis and diagnosis. The consequences of erythrocytosis — hyperviscosity, thrombosis, and bleeding — dominate the clinical picture. Over time, PV may transform to post-PV myelofibrosis or, less commonly, to blast-phase (acute myeloid leukemia), conferring a worsened prognosis.
2. Epidemiology
PV has an estimated incidence of 0.4–2.8 cases per 100,000 persons per year in Western populations. Prevalence is approximately 22 per 100,000. The disease predominantly affects older adults, with a median age at diagnosis of 60–65 years, though cases in younger patients (<40 years) are well documented and carry distinct management considerations. There is a slight male predominance (male-to-female ratio approximately 1.2:1), though some registry data show near-equal sex distribution. Ashkenazi Jewish populations appear to have a higher incidence, suggesting possible genetic predisposition. PV is rare in children and adolescents. Geographic variation is recognized but may partly reflect differences in diagnostic practices.
3. Pathophysiology
JAK2 V617F Mutation and JAK-STAT Signaling
The JAK2 V617F somatic point mutation (valine-to-phenylalanine substitution at position 617 in the pseudokinase domain of Janus kinase 2) is present in approximately 96–99% of PV patients. The mutation constitutively activates JAK2 kinase by disrupting autoinhibition, leading to continuous phosphorylation of downstream STAT3 and STAT5 transcription factors independent of normal cytokine receptor engagement (erythropoietin receptor, thrombopoietin receptor, G-CSF receptor). This results in cytokine-hypersensitive and ultimately cytokine-independent proliferation and survival of erythroid progenitors.
The remaining ~4% of PV cases harbor mutations in exon 12 of JAK2, which produce a predominantly erythroid phenotype with lower leukocyte counts and a lower likelihood of concomitant thrombocytosis. Additional somatic mutations in TET2, DNMT3A, ASXL1, SF3B1, and SRSF2 are detected in a proportion of patients and may influence disease phenotype, progression risk, and leukemic transformation risk.
Consequences of Erythrocytosis
Erythrocytosis increases whole-blood viscosity exponentially as the hematocrit rises above 45–50%. Increased viscosity impairs microvascular flow, promotes stasis, and activates coagulation — a state of Virchow's triad that predisposes to both arterial and venous thrombosis. Paradoxically, extreme thrombocytosis in some patients causes acquired von Willebrand syndrome (loss of high-molecular-weight VWF multimers), predisposing to bleeding. Splenomegaly from extramedullary hematopoiesis contributes to hypersplenism, anemia, and abdominal symptoms.
4. Etiology and Risk Factors
- JAK2 V617F somatic mutation: The primary driver in ~97% of cases; arises in a hematopoietic stem cell.
- JAK2 exon 12 mutations: Account for ~3% of cases; predominantly erythroid phenotype.
- Age: Incidence rises markedly after age 50; median diagnosis at 60–65 years.
- Familial predisposition: A small fraction of MPNs cluster in families; germline JAK2 haplotype 46/1 ("GGCC haplotype") increases susceptibility to somatic JAK2 mutation.
- Prior radiation exposure: Occupational or therapeutic ionizing radiation has been associated with increased MPN risk.
- Benzene exposure: Chronic exposure to benzene-containing solvents is a recognized risk factor for myeloid malignancies including MPNs.
- Clonal hematopoiesis of indeterminate potential (CHIP): Age-related acquisition of somatic mutations including JAK2 V617F in a small hematopoietic clone may precede overt PV.
5. Clinical Presentation
Symptoms
Many patients are asymptomatic at diagnosis, identified incidentally from a routine blood count. Symptomatic patients most commonly present with:
- Aquagenic pruritus: Intense, burning itch provoked by warm water contact; caused by mast cell degranulation and elevated histamine; present in 40–70% of patients and highly characteristic of PV.
- Headache, dizziness, visual disturbances, and tinnitus: Manifestations of cerebral hyperviscosity and microvascular occlusion.
- Erythromelalgia: Episodic burning pain and erythema of the extremities (especially toes and feet) caused by platelet-mediated microvascular occlusion; responds dramatically to aspirin.
- Plethora (facial ruddiness): Ruddy cyanosis of the face and mucous membranes.
- Fatigue and impaired concentration: Highly prevalent constitutional symptoms.
- Splenomegaly: Present in ~40–70% at diagnosis; causes early satiety, left upper quadrant fullness, and pain.
- Gout: Hyperuricemia from accelerated cell turnover precipitates gout and nephrolithiasis.
Thrombosis and Bleeding
Thrombosis occurs in 15–30% of patients at or before diagnosis and is the leading cause of morbidity and mortality. Both arterial (stroke, TIA, myocardial infarction, peripheral arterial occlusion) and venous (deep vein thrombosis, pulmonary embolism, splanchnic vein thrombosis — Budd-Chiari syndrome, portal vein thrombosis) events occur. Splanchnic vein thrombosis in a young person should prompt JAK2 testing even if blood counts appear normal. Bleeding complications (gastrointestinal, skin) occur less commonly, often in the setting of extreme thrombocytosis with acquired VWF syndrome.
6. Diagnosis
WHO 2022 Diagnostic Criteria
Diagnosis requires meeting all three major criteria OR the first two major criteria plus the minor criterion:
Major criteria:
- Hemoglobin >16.5 g/dL in men / >16.0 g/dL in women, or hematocrit >49% in men / >48% in women, or increased red cell mass (>25% above mean normal predicted value).
- Bone marrow biopsy showing hypercellularity for age with trilineage (panmyelosis) proliferation — pleomorphic mature megakaryocytes.
- Presence of JAK2 V617F or JAK2 exon 12 mutation.
Minor criterion:
- Subnormal serum erythropoietin (EPO) level.
Laboratory Evaluation
- Complete blood count (CBC): Elevated hemoglobin, hematocrit, and often elevated leukocyte count (neutrophilia) and platelet count.
- Serum EPO level: Suppressed (<3.6 mU/mL) in PV; distinguishes from secondary erythrocytosis (where EPO is elevated or normal).
- JAK2 V617F allele-specific PCR or next-generation sequencing: Positive in ~97% of PV; mandatory for diagnosis.
- JAK2 exon 12 sequencing: If V617F negative, particularly in isolated erythrocytosis.
- Bone marrow aspirate and trephine biopsy: Panmyelosis with pleomorphic megakaryocytes; reticulin fibrosis graded per WHO (MF-0 to MF-3).
- Uric acid, LDH: Commonly elevated due to increased cell turnover.
- Vitamin B12 and binding capacity: May be elevated from granulocyte-derived transcobalamin.
- Pulse oximetry / arterial blood gas: To exclude hypoxia-driven secondary erythrocytosis.
- Abdominal ultrasound: To assess spleen size and exclude splanchnic vein thrombosis.
7. Treatment
Risk Stratification
All patients require cytoreduction-independent interventions; cytoreductive therapy is reserved for higher-risk patients:
- Low risk: Age <60 years AND no prior thrombotic event.
- High risk: Age ≥60 years OR prior thrombotic event.
All Patients
- Phlebotomy (venesection): Target hematocrit <45% in men and <42% in women (CYTO-PV trial demonstrated that hematocrit <45% significantly reduced cardiovascular death and major thrombosis compared to 45–50%). Each session removes 250–500 mL of whole blood.
- Low-dose aspirin (81–100 mg/day): Reduces microvascular events (erythromelalgia, TIA) and thrombosis risk, particularly for JAK2 V617F-positive PV; demonstrated benefit in the ECLAP trial. Avoid if platelet count >1,000–1,500 × 10⁹/L due to acquired VWF syndrome.
- Cardiovascular risk factor modification: Aggressive management of hypertension, diabetes, dyslipidemia, and smoking cessation.
High-Risk Patients: Cytoreductive Therapy
- Hydroxyurea (hydroxycarbamide): First-line cytoreductive agent; inhibits ribonucleotide reductase, reducing proliferation of all myeloid lineages. Typical dose 500–2000 mg/day orally; well-tolerated; low leukemogenic potential at standard doses. Monitor CBC weekly initially.
- Ruxolitinib (Jakafi): A JAK1/JAK2 inhibitor; approved second-line for patients resistant to or intolerant of hydroxyurea. The RESPONSE trial demonstrated superiority over best available therapy in achieving hematocrit control and spleen volume reduction. Starting dose 10 mg twice daily; titrate based on response.
- Interferon-alfa (pegylated, e.g., ropeginterferon alfa-2b [Besremi]): Reduces JAK2 V617F allele burden and may induce molecular remission; preferred in younger patients and those of childbearing potential. Ropeginterferon alfa-2b is FDA-approved (2021) for PV.
- Busulfan: Oral alkylating agent used in older patients or where other agents are contraindicated; leukemogenic risk with prolonged use.
- Radioactive phosphorus (³²P): Largely replaced by modern cytoreductive agents; occasionally used in elderly patients.
8. Complications
- Arterial and venous thrombosis: Leading cause of death in PV; includes stroke, MI, DVT, PE, Budd-Chiari syndrome.
- Transformation to post-PV myelofibrosis (PPV-MF): Occurs in ~10–20% of patients at 10 years and up to 25–35% at 20 years; presents with progressive anemia, splenomegaly, and constitutional symptoms.
- Blast-phase transformation (post-PV AML): Occurs in ~2–5% of patients at 10 years; prognosis is extremely poor (median survival <6 months); risk increases with prior alkylating agent therapy.
- Major hemorrhage: GI bleeding, intracranial hemorrhage; more common in patients with extreme thrombocytosis and acquired VWF syndrome.
- Gout and renal calculi: From hyperuricemia due to increased cell turnover; managed with allopurinol.
- Pruritus-related quality of life impairment: Severe aquagenic pruritus significantly reduces quality of life.
9. Prognosis
PV has a significantly shorter life expectancy compared to an age-matched general population, primarily due to thrombotic events and disease transformation. Median overall survival in contemporary series is approximately 14–19 years from diagnosis. The IPSET-thrombosis score integrates age, cardiovascular risk factors, thrombotic history, and JAK2 V617F status to stratify thrombotic risk. Adverse prognostic factors include older age, leukocytosis (WBC >15 × 10⁹/L), abnormal karyotype, and additional somatic mutations (particularly SRSF2, IDH1/2, RUNX1).
The MIPSS-PV score (Mutation-Enhanced International Prognostic Score System) incorporates molecular data into prognostication. Patients with low-risk features and good hematocrit control can have near-normal life expectancy; those who transform to AML have a dismal prognosis.
10. Prevention
- Hematocrit control below 45%: The single most important modifiable risk factor for thrombosis prevention; achieved via phlebotomy and/or cytoreductive therapy.
- Antiplatelet therapy: Low-dose aspirin reduces microvascular thrombotic events in all PV patients without contraindications.
- Cytoreductive therapy in high-risk patients: Hydroxyurea or interferon-alfa reduces thrombotic risk and disease burden.
- Anticoagulation: Long-term anticoagulation (vitamin K antagonists or direct oral anticoagulants) is indicated after venous thromboembolism, with hematocrit control maintained concomitantly.
- Cardiovascular risk reduction: Smoking cessation, blood pressure control, and management of dyslipidemia reduce thrombotic event rates.
- Regular monitoring: CBC every 3 months; bone marrow reassessment if clinical deterioration or progressive cytopenias.
11. Recent Research and Advances
The approval of ropeginterferon alfa-2b (Besremi) by the FDA in 2021 marked a significant milestone, offering a disease-modifying option capable of reducing JAK2 V617F allele burden and achieving molecular responses not seen with hydroxyurea. The PROUD-PV and CONTINUATION-PV trials demonstrated its superiority to hydroxyurea in molecular response rates. Ruxolitinib remains the validated second-line option after the RESPONSE and RESPONSE-2 trials established its efficacy in hydroxyurea-resistant/intolerant patients.
Emerging targets include calreticulin (CALR) mutations in JAK2-negative cases; novel JAK2-specific inhibitors are in clinical development to overcome the limitations of ruxolitinib (incomplete JAK2 selectivity, immunosuppression). Hepcidin mimetics (rusfertide) are in Phase 3 trials (VERIFY trial) to reduce iron-restricted erythropoiesis and decrease phlebotomy requirements. The biology of clonal evolution and the role of additional somatic mutations in disease progression and leukemic transformation are active areas of research. Patient-reported outcomes, particularly aquagenic pruritus and fatigue measurement using the MPN-SAF TSS instrument, are increasingly incorporated into trial endpoints.
Research Papers
The following PubMed topic searches return current peer-reviewed literature relevant to this condition. Each link opens a live PubMed query.
- Polycythemia vera JAK2
- Polycythemia vera diagnosis
- Polycythemia vera phlebotomy
- Hydroxyurea polycythemia vera
- Ruxolitinib polycythemia vera
- Polycythemia vera thrombosis
- Myeloproliferative neoplasms WHO classification
- Polycythemia vera myelofibrosis transformation
- Interferon alpha polycythemia vera
- Polycythemia vera guidelines
- Secondary erythrocytosis
- Polycythemia vera survival
Connections
- Hematology
- Anemia
- Deep Vein Thrombosis
- Stroke
- Complete Blood Count
- Hemochromatosis
- Thrombocytopenia
- Atrial Fibrillation
- Gout
- Iron
- Leukemia
- Hypertension
- Uric Acid
- Pulmonary Embolism
- Disseminated Intravascular Coagulation
- Sickle Cell Disease
- Von Willebrand Disease
- Hemophilia