Von Willebrand Disease
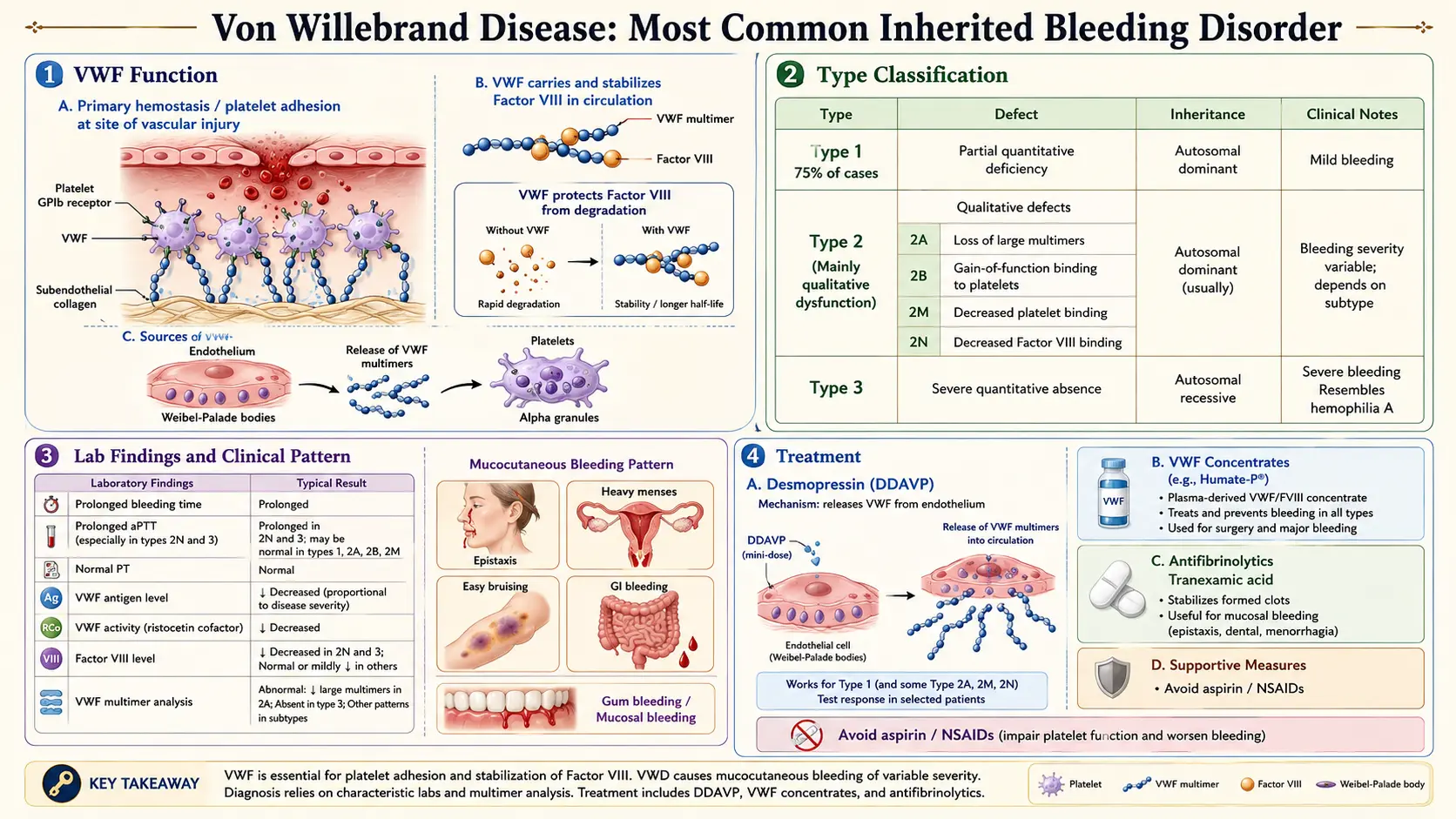
Table of Contents
- Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Recent Research and Advances
- Research Papers
- Connections
- Featured Videos
1. Overview
Von Willebrand disease (VWD) is the most common inherited bleeding disorder in humans, caused by quantitative deficiency or qualitative dysfunction of von Willebrand factor (VWF) — a large multimeric glycoprotein essential for both primary hemostasis (platelet adhesion at sites of vascular injury) and secondary hemostasis (carrier and stabilizer of coagulation Factor VIII). First described by Finnish physician Erik Adolf von Willebrand in 1926, the disorder was initially called "hereditary pseudohemophilia."
VWD is classified by the International Society on Thrombosis and Haemostasis (ISTH) into three main types: Type 1 (partial quantitative deficiency, ~70–80% of cases), Type 2 (qualitative defects, ~20–25%), and Type 3 (virtual complete absence, ~1–5%). The bleeding phenotype is predominantly mucocutaneous — epistaxis, menorrhagia, easy bruising, and excessive bleeding with dental procedures or surgery — distinguishing it from the joint-dominant pattern of hemophilia.
2. Epidemiology
VWD is estimated to affect approximately 1% of the general population when all mild forms are included, making it the most prevalent inherited coagulopathy worldwide. Symptomatic VWD (with significant bleeding symptoms) has an estimated prevalence of approximately 100–125 per million persons. Type 1 VWD, inherited in an autosomal dominant pattern, is the most common form; penetrance and expressivity vary considerably even within families. Types 2A, 2B, 2M, and 2N have specific patterns of inheritance (predominantly autosomal dominant, except 2N which is recessive). Type 3 VWD, the most severe form, is autosomal recessive with an estimated prevalence of 0.5–5 per million.
VWD affects both sexes equally in terms of gene frequency; however, women are disproportionately symptomatic due to menorrhagia and obstetric hemorrhage. VWD is detected across all ethnic groups, though prevalence data vary by study methodology and diagnostic thresholds used.
3. Pathophysiology
Von Willebrand Factor Structure and Function
VWF is encoded by the VWF gene on chromosome 12p13.3 and is synthesized as a 2,813 amino acid pre-pro-peptide in endothelial cells and megakaryocytes. After processing and multimerization in the endoplasmic reticulum and Golgi apparatus, VWF is stored in endothelial Weibel-Palade bodies and platelet alpha-granules, from which it is released upon stimulation (thrombin, DDAVP, epinephrine). VWF circulates as multimers ranging from dimers to ultra-large multimers (>10,000 kDa); the ultra-large high-molecular-weight (HMW) multimers are most hemeostatically effective.
VWF performs two critical functions:
- Primary hemostasis: Under high shear conditions, VWF unfolds and its A1 domain binds platelet surface glycoprotein Ib (GPIb), anchoring platelets to subendothelial collagen (via VWF A3 domain) at sites of vascular injury. This tethering allows subsequent platelet activation and GPIIb/IIIa-mediated aggregation.
- Secondary hemostasis: VWF binds and protects Factor VIII (via its D′D3 domain) from premature proteolytic degradation by Factor Xa and activated protein C in the circulation, prolonging FVIII half-life approximately 3-fold. Deficiency of VWF therefore causes secondary FVIII deficiency (FVIII levels 1–30% in Type 3 VWD).
VWF multimer size is regulated by the metalloprotease ADAMTS13 (a disintegrin and metalloprotease with thrombospondin motifs, member 13), which cleaves ultra-large VWF multimers in the bloodstream. Deficiency of ADAMTS13 causes TTP; conversely, Type 2A and 2B VWD result from gain-of-function VWF mutations that increase susceptibility to ADAMTS13 proteolysis or spontaneous platelet binding, respectively.
Classification and Genetic Mechanisms
- Type 1 VWD: Heterozygous loss-of-function mutations in VWF cause partial quantitative deficiency (VWF:Ag typically 20–50 IU/dL). Pathogenic variants include missense mutations causing intracellular retention, secretion defects, or accelerated VWF clearance.
- Type 2A: Qualitative defect with loss of HMW multimers; mutations cause increased ADAMTS13 proteolysis or impaired multimer assembly (gain-of-function or dominant-negative effects in VWF A2 domain).
- Type 2B: Gain-of-function mutations in VWF A1 domain cause spontaneous (shear-independent) platelet binding, leading to loss of HMW multimers and thrombocytopenia (platelet clearance with VWF).
- Type 2M: Qualitative defect with preserved multimer distribution but decreased platelet-dependent function (mutations in A1 domain reducing GPIb binding affinity).
- Type 2N (Normandy): Mutations in VWF D′D3 domain markedly reduce FVIII binding affinity, causing secondary FVIII deficiency mimicking mild Hemophilia A; autosomal recessive.
- Type 3: Homozygous or compound heterozygous null mutations causing near-complete absence of VWF; severe bleeding phenotype with FVIII levels 1–10%.
4. Etiology and Risk Factors
- Inherited VWF gene mutations: The primary cause; >500 distinct VWF variants have been catalogued in the ISTH VWF Database.
- Autosomal dominant inheritance (Types 1, 2A, 2B, 2M): A single mutant allele is sufficient to cause disease; expressivity is highly variable.
- Autosomal recessive inheritance (Types 2N, 3): Both alleles must carry pathogenic variants; consanguinity increases risk for Type 3.
- Blood group O: Individuals with blood group O have VWF levels ~25% lower than non-O individuals due to increased ADAMTS13-mediated and non-ADAMTS13 clearance; this is a continuous quantitative trait modifier, not a disease per se, but can confound diagnosis and lower the diagnostic VWF threshold for blood group O individuals.
- Acquired von Willebrand syndrome (AVWS): Not a genetic disorder; results from autoantibodies against VWF, increased shear stress-induced VWF degradation (aortic stenosis — Heyde syndrome; ventricular assist devices; ECMO), or adsorption of VWF onto tumor cells (Wilms tumor, lymphomas); managed by treating the underlying condition.
5. Clinical Presentation
Bleeding Patterns
VWD produces a predominantly mucocutaneous bleeding phenotype, reflecting impaired primary hemostasis:
- Epistaxis: The most common symptom; recurrent, often bilateral, frequently requiring cauterization or packing.
- Menorrhagia: Heavy menstrual bleeding (HMB) is the presenting symptom in ~70–80% of symptomatic women with VWD; defined as >80 mL per cycle or bleeding lasting >7 days; commonly causes iron deficiency anemia.
- Easy bruising: Particularly over bony prominences; bruises disproportionate to trauma.
- Prolonged bleeding from cuts and minor wounds.
- Excessive bleeding with dental extraction, tonsillectomy, or surgery: Often the first recognized manifestation.
- Postpartum hemorrhage: Primary (within 24 hours) or secondary (24 hours to 6 weeks); VWF levels rise during pregnancy (hormonal effect), which may mask VWD, followed by a postpartum drop back to baseline.
- GI bleeding: More common in Type 2A and Type 3; arteriovenous malformations of the GI tract (angiodysplasia) are associated with VWD, particularly Type 2A and Type 3, possibly due to shear-dependent VWF-mediated angiogenesis signaling.
- Hemarthrosis and muscle hematomas: Uncommon; occur in Type 3 (FVIII <10%) and Type 2N.
Severity Classification
Bleeding severity in VWD correlates with VWF activity levels and subtype:
- Mild: Type 1 (VWF 20–50 IU/dL); mucocutaneous symptoms; often identified after challenge (surgery, dental).
- Moderate: Type 2 subtypes and Type 1 with lower VWF levels; more prominent mucocutaneous symptoms.
- Severe: Type 3 (VWF <3 IU/dL); resembles moderate hemophilia A with hemarthroses possible.
6. Diagnosis
Initial Screening
- aPTT: May be mildly prolonged (due to secondary FVIII deficiency) in Types 1, 2N, and 3; may be normal in mild Type 1.
- PT: Normal.
- Platelet count: Normal in most types; may be mildly reduced in Type 2B.
- PFA-100 (platelet function analyzer): Closure time prolonged; a sensitive screen for VWD and platelet disorders, though non-specific.
- Bleeding time: Historically used; largely replaced by PFA-100 due to poor reproducibility.
Specific VWD Assays (ISTH-SSC Recommended)
- VWF antigen (VWF:Ag): Immunoassay measuring total VWF protein concentration; reduced in Types 1 and 3; may be normal or low-normal in Type 2.
- VWF ristocetin cofactor activity (VWF:RCo): Functional assay measuring VWF-GPIb interaction; reduced in Types 1, 2A, 2B, 2M; the traditional functional assay, now largely replaced by GPIb-binding assays.
- VWF GPIb-binding activity (VWF:GPIbM or VWF:GPIbR): More reproducible modern functional assays replacing VWF:RCo in many centers.
- VWF:RCo/VWF:Ag or VWF:GPIbM/VWF:Ag ratio: Ratio <0.6 suggests Type 2A or 2M (qualitative defect with loss of HMW multimers); ratio normal in Type 1 and 2N.
- FVIII activity (FVIII:C): Reduced in Type 3 and Type 2N; mildly reduced in Type 1.
- VWF:FVIII binding (VWF:FVIIIB): Specifically detects Type 2N; markedly reduced FVIII binding capacity.
- Ristocetin-induced platelet aggregation (RIPA): Low-dose ristocetin (<0.6 mg/mL) causes platelet aggregation in Type 2B (gain-of-function); absent or reduced at standard doses in Type 2M and 2A.
- VWF multimer analysis (gel electrophoresis): Absent HMW multimers in Types 2A and 2B; uniform reduction in Type 1; absent all multimers in Type 3.
- VWF gene sequencing: Confirms subtype, identifies pathogenic variant, enables family screening; especially important for Type 2 subtyping and Type 2N vs. Hemophilia A differentiation.
Note: Repeat testing is important, as VWF levels are affected by blood group, inflammation, stress, hormonal status, thyroid function, and age. ISTH guidelines recommend at least two abnormal results on different occasions for diagnosis.
7. Treatment
DDAVP (Desmopressin; 1-desamino-8-D-arginine vasopressin)
DDAVP is the treatment of choice for mild-to-moderate VWD when effective. It releases endogenous VWF (and FVIII) from endothelial Weibel-Palade bodies, acutely raising VWF and FVIII levels 2–5-fold for 4–8 hours. Formulations include:
- Intravenous DDAVP (0.3 µg/kg): Most rapid and consistent rise in VWF.
- Intranasal DDAVP (Stimate; 150 µg/actuation): One spray (<50 kg) or two sprays (>50 kg); convenient for outpatient use.
Efficacy must be confirmed by a pre/post DDAVP challenge test measuring VWF and FVIII levels. DDAVP is effective in most Type 1 patients. It is contraindicated in Type 2B (may worsen thrombocytopenia by releasing ultra-large VWF, promoting platelet aggregation) and ineffective in Type 3. Tachyphylaxis occurs with repeat dosing every 24 hours. Side effects include facial flushing, headache, and hyponatremia (restrict free water intake for 24 hours after dosing; avoid in cardiovascular disease).
VWF Replacement Therapy
- Plasma-derived VWF/FVIII concentrates (e.g., Humate-P, Wilate, Alphanate): Contain VWF and FVIII; indicated for types not responsive to DDAVP, Type 3, and major surgery/severe bleeding. Dosing based on VWF:RCo or GPIbM units; target VWF:RCo >100 IU/dL for major surgery, >50 IU/dL for minor surgery, and >30 IU/dL for prophylaxis.
- Recombinant VWF (vonicog alfa; Vonvendi): FDA-approved for adults with VWD; produced in CHO cells; contains VWF without FVIII; may be given with recombinant FVIII initially for FVIII deficit correction.
Adjunctive Therapies
- Tranexamic acid (15–25 mg/kg orally/IV TID) and epsilon-aminocaproic acid: Antifibrinolytics that stabilize clots by inhibiting plasminogen activators; particularly effective for mucosal bleeds, dental procedures, and menorrhagia; may be used alone in mild VWD or as adjuncts.
- Combined oral contraceptive pills: Increase VWF and FVIII levels hormonally; highly effective for menorrhagia in Type 1 VWD.
- Progestogen-releasing intrauterine device (levonorgestrel IUD; Mirena): Reduces menstrual blood loss; does not raise VWF levels but dramatically reduces menstrual flow.
- Iron supplementation: For iron deficiency anemia secondary to chronic mucosal blood loss.
8. Complications
- Iron deficiency anemia: Chronic blood loss from menorrhagia, GI bleeding, or epistaxis; often severe in Type 3.
- Hemarthrosis and hemophilic arthropathy: Uncommon; seen in Type 3 VWD and Type 2N with FVIII <10%; may require joint replacement if advanced.
- Postpartum hemorrhage: VWF levels rise during pregnancy but fall postpartum; highest risk in Type 3 and Type 2; requires careful peripartum planning with VWF replacement.
- Gastrointestinal angiodysplasia: Particularly problematic in Types 2A and 3; recurrent GI bleeding; VWF HMW multimers play a role in suppressing angiogenesis of intestinal microvasculature.
- Inhibitor development: Alloantibodies against VWF in 5–10% of Type 3 patients following VWF replacement; can cause anaphylaxis with subsequent VWF infusions.
- Misdiagnosis and delayed diagnosis: VWD, particularly in women, is often attributed to "normal heavy periods" or anxiety; average diagnostic delay can exceed 10 years.
9. Prognosis
For the vast majority of patients with VWD — particularly Type 1 — prognosis is excellent with appropriate diagnosis and management. Life expectancy is normal. Quality of life is the primary concern, particularly for women with menorrhagia and patients with severe Type 3. Effective DDAVP or VWF replacement therapy enables patients to undergo surgery and manage bleeding episodes without life-threatening hemorrhage. Type 3 patients with inhibitors face the most challenging management, analogous to hemophilia with inhibitors. Early diagnosis, especially in women and children, dramatically reduces morbidity from uncontrolled bleeding and unnecessary iron deficiency.
10. Prevention
- Genetic counseling: Family screening of first-degree relatives after index case diagnosis; prenatal diagnosis available for Type 3 (severe fetal bleeding risk at delivery).
- Pre-procedural planning: DDAVP or VWF replacement administered before any elective surgery, dental procedures, or invasive procedures; hematology consultation for perioperative management.
- Avoidance of antiplatelet agents: NSAIDs and aspirin impair platelet function and worsen bleeding; acetaminophen preferred.
- Medical alert identification: Patients should carry medical alert information specifying their VWD type, treatment agent, and emergency contact.
- Comprehensive care at hemophilia treatment centers: Multidisciplinary teams (hematology, gynecology, physical therapy) optimize management and reduce complications.
- Hormonal management: Pre-emptive hormonal therapy in women with menorrhagia prevents iron deficiency and reduces emergency bleeding events.
11. Recent Research and Advances
The approval of recombinant VWF (vonicog alfa; Vonvendi) in 2015 (USA) provided the first recombinant VWF product, eliminating plasma-derived infection risk and enabling precise VWF dosing without concurrent FVIII loading. Clinical trials are evaluating vonicog alfa for prophylaxis in severe VWD. Fitusiran and other rebalancing strategies targeting natural anticoagulant pathways are under investigation for VWD, particularly Type 3 with FVIII deficiency.
ISTH updated its diagnostic criteria and assay recommendations in 2021, establishing VWF:GPIbM (VWF GPIb-binding using mutant GPIb) as the preferred functional assay, replacing the historically problematic VWF:RCo assay. The International VWD Outcome Study (i-WAS) and the Zimmermann Willebrand Investigators (ZWI-SS) cohorts are generating large-scale real-world evidence on bleeding severity, treatment efficacy, and quality of life outcomes. Molecular epidemiology studies continue to characterize the full spectrum of VWF variants, particularly those causing reduced VWF clearance (protective variants) vs. increased clearance, with the latter identified as an important mechanism in severe Type 1 VWD.
The role of ABO blood group in VWF biology has been further clarified: Group O individuals have VWF levels 25–30% lower than non-O, and the ISTH now recommends blood group-specific reference ranges. Emerging evidence supports a role for VWF in angiogenesis regulation, with implications for understanding GI angiodysplasia in VWD.
Research Papers
The following PubMed topic searches return current peer-reviewed literature relevant to this condition. Each link opens a live PubMed query.
- Von Willebrand disease classification
- Von Willebrand factor laboratory
- Von Willebrand disease type 1
- Von Willebrand disease type 2
- Von Willebrand disease type 3
- Desmopressin DDAVP von Willebrand
- Von Willebrand factor concentrate
- Acquired von Willebrand syndrome
- Von Willebrand disease diagnosis
- Von Willebrand disease guidelines
- Von Willebrand disease pregnancy
- Heavy menstrual bleeding von Willebrand
Connections
- Hematology
- Hemophilia
- Thrombocytopenia
- Anemia
- Disseminated Intravascular Coagulation
- Iron
- Vitamin K
- Complete Blood Count
- Iron Deficiency Anemia
- Deep Vein Thrombosis
- Hemochromatosis
- Polycythemia Vera
- Sickle Cell Disease
- Thalassemia
- Headache
- Anxiety
- Coagulation Panel