Vitamin B12 for Nerve Health and Neuropathy
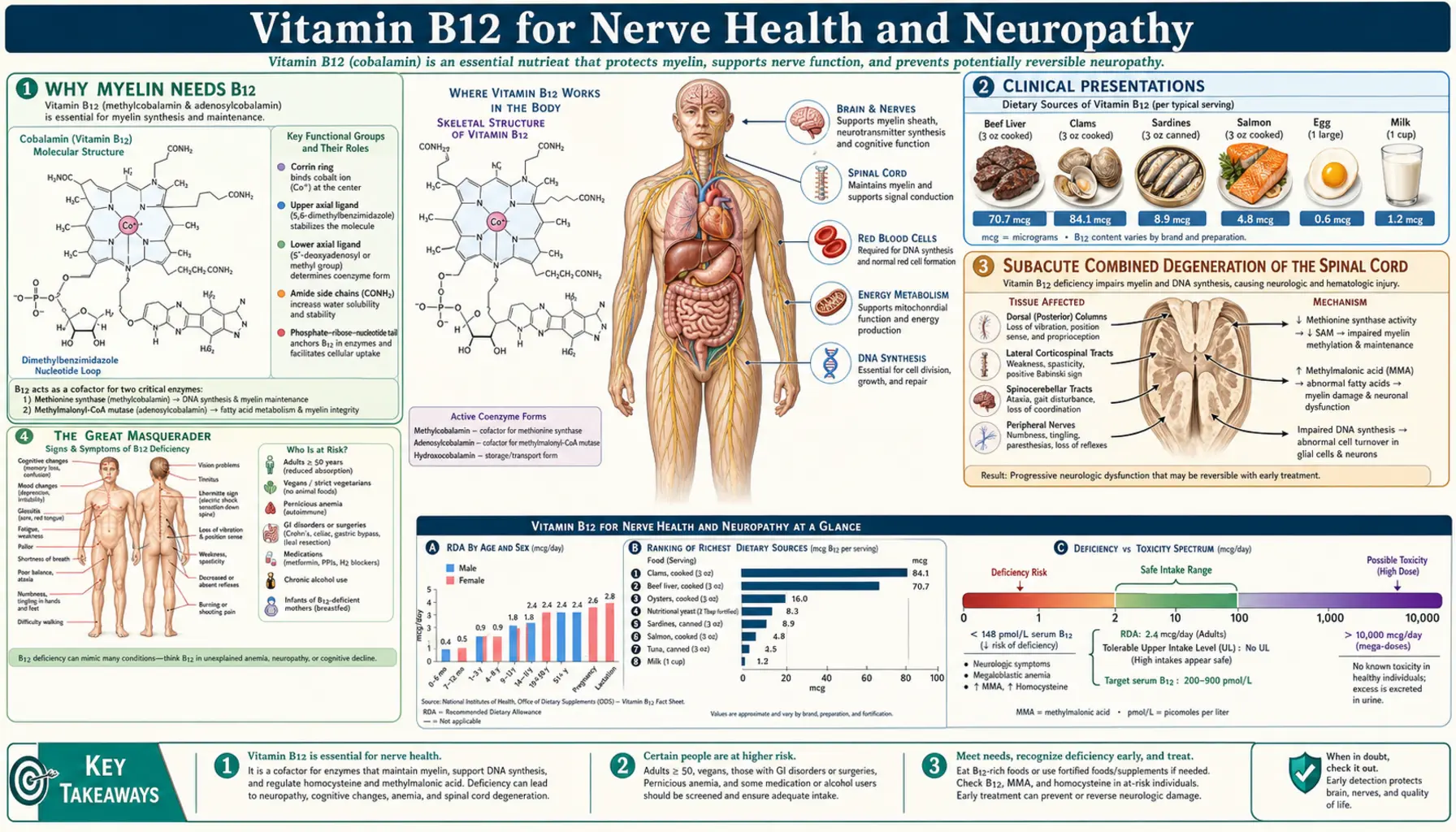
B12 deficiency is the great masquerader of clinical neurology. It mimics multiple sclerosis, dementia, depression, diabetic neuropathy, and chronic fatigue with such convincing fidelity that it is misdiagnosed in tens of thousands of patients every year. The cruelest feature of B12 deficiency is that the damage to myelin and nerve tissue can become permanent if the deficiency persists past a certain threshold — making early recognition and aggressive repletion one of the highest-stakes interventions in medicine. This deep-dive walks through the myelin synthesis mechanism, the classic stocking-glove peripheral neuropathy presentation, subacute combined degeneration of the spinal cord, why methylmalonic acid (MMA) catches deficiency long before serum B12 does, and the high-dose methylcobalamin protocols that integrative neurologists use to maximize recovery.
Interactive Visualization Vitamin B12’s Impossible Journey Five stages, two carrier proteins, a working stomach and one specific stretch of gut. Break any link — a PPI, metformin, pernicious anaemia — and absorption collapses. Launch →
Table of Contents
- Why Myelin Needs B12
- Clinical Presentations
- Subacute Combined Degeneration
- The Great Masquerader
- MMA as the Definitive Marker
- Methylcobalamin vs Cyanocobalamin
- High-Dose 5000 mcg Sublingual Protocol
- Recovery Timeline and What to Expect
- Combination Strategies
- Cautions
- Key Research Papers
- Connections
- Featured Videos
Why Myelin Needs B12
Myelin is the fatty, multi-layered insulating sheath that wraps every nerve fiber in the central and peripheral nervous systems. It allows nerve impulses to travel at speeds of up to 100 meters per second through a mechanism called saltatory conduction — the electrical signal "jumps" from one node of Ranvier to the next instead of having to propagate continuously along the entire axon membrane. Without intact myelin, conduction velocity collapses, signals are degraded or lost entirely, and the nerve becomes electrically dysfunctional even if the axon itself remains anatomically intact.
Myelin is composed primarily of two lipid molecules: phosphatidylcholine and sphingomyelin. Both require methyl groups for their synthesis, and the universal donor of methyl groups in mammalian biology is S-adenosylmethionine (SAMe). SAMe is produced from methionine, which in turn is produced when homocysteine is remethylated by the enzyme methionine synthase. Methionine synthase has only one cofactor: methylcobalamin, the methylated active form of vitamin B12.
The chain of dependence is therefore: B12 (methylcobalamin) → methionine synthase activity → methionine → SAMe → phospholipid methylation → myelin synthesis and repair. Break any link in this chain — most commonly the first one, B12 sufficiency — and myelin synthesis falters. Because nerves are constantly turning over their myelin, the effect of B12 deficiency is progressive demyelination. Surviving axons continue to function, but progressively more slowly and unreliably, until eventually the axon itself dies and the deficit becomes permanent.
This is why B12 deficiency produces neurological symptoms that look so different from the symptoms of other vitamin deficiencies. The defect is not in the metabolic activity of the nerve cell; it is in the structural integrity of its insulation. Recovery is possible if myelin can be rebuilt, but only up to the point where the underlying axon has been preserved.
Clinical Presentations
B12-related nerve dysfunction can present in any of several patterns, often overlapping:
- Distal symmetric peripheral neuropathy — the most common presentation. Stocking-glove distribution of numbness, tingling, "pins and needles," burning, and electric-shock pain starting in the toes and feet and gradually ascending. Worse at night. The presentation is clinically indistinguishable from diabetic peripheral neuropathy, which is one reason B12 status should be checked in every diabetic patient with neuropathic symptoms before assuming the diabetes is the cause.
- Sensory ataxia and balance loss — degeneration of the dorsal columns of the spinal cord interrupts proprioceptive signaling. Patients can't sense the position of their feet without looking, gait becomes wide-based and unsteady, and falls in the dark become common. The Romberg test (close eyes while standing with feet together) becomes positive — the patient sways or falls. This pattern is the hallmark of B12-related subacute combined degeneration.
- Motor weakness and spasticity — degeneration of the lateral columns (corticospinal tracts) produces upper-motor-neuron signs: leg stiffness, hyperreflexia, Babinski sign, and weakness. In late disease, paraplegia can develop.
- Optic neuropathy — the optic nerve is exquisitely sensitive to B12 deficiency. Painless progressive bilateral visual loss with central scotoma and color-vision defect (especially red-green) is the classic presentation. Severe untreated cases progress to optic atrophy and permanent blindness.
- Autonomic neuropathy — orthostatic hypotension, gastroparesis, urinary urgency or incontinence, erectile dysfunction, and abnormal sweating. Frequently overlaps with somatic neuropathy.
- Lhermitte's sign — an electric-shock sensation that shoots down the spine and into the limbs when the neck is flexed. Classically associated with multiple sclerosis but also a feature of B12-related dorsal column disease.
What unites all of these presentations is that they can occur before any hematological abnormality is detectable. Lindenbaum's landmark 1988 NEJM paper documented dozens of patients with biopsy-proven B12-deficient neurological disease who had completely normal hemoglobin, MCV, and CBC. This decoupling of neurological and hematological deficiency is one of the most clinically important features of B12: a normal CBC does not rule out B12 deficiency.
Subacute Combined Degeneration of the Spinal Cord
Subacute combined degeneration (SCD) is the textbook neurological complication of severe, prolonged B12 deficiency. The "combined" in the name refers to the simultaneous involvement of multiple spinal cord tracts: the dorsal columns (carrying proprioception, vibration sense, and fine touch from the body to the brain) and the lateral corticospinal tracts (carrying motor commands from the brain to the body). Both tracts demyelinate, and eventually axons degenerate, producing a mixed sensory-motor pattern that progresses from the feet upward.
The classic SCD patient has:
- Loss of vibration sense and proprioception in the feet, ascending to the knees and beyond
- Positive Romberg sign (loss of balance when eyes close)
- Wide-based, unsteady gait that gets worse in the dark
- Spasticity, hyperreflexia, and a positive Babinski reflex (upper-motor-neuron signs from corticospinal tract involvement)
- Weakness in the legs
- Frequently, peripheral nerve involvement on top of the spinal cord findings (mixed picture)
SCD is what made B12 deficiency such a feared diagnosis before the cause was understood. In the era before B12 was identified (deficiency was called "pernicious" anemia precisely because it was uniformly fatal), patients with pernicious anemia would progress to SCD with paraplegia, and many would die from complications of progressive neurological disability. Once the cause was identified in 1934, parenteral B12 became the treatment, and full neurological recovery became possible — if treatment began before axonal damage was too advanced.
The critical clinical principle for SCD: damage is reversible during the demyelination phase and irreversible after axonal loss. The window for full recovery is approximately 6-12 months from symptom onset. Beyond that, partial recovery is still possible with aggressive treatment but complete reversal becomes unlikely. Patients presenting with SCD-pattern symptoms should be on parenteral hydroxocobalamin or methylcobalamin within 24 hours of suspected diagnosis — do not wait for serum B12 results, do not wait for the appointment with the neurologist, do not wait for the MRI. Start treatment, then complete the workup.
The Great Masquerader
B12 deficiency is called the great masquerader because the spectrum of presentations overlaps so completely with so many other conditions. A 70-year-old presenting with progressive memory loss, gait unsteadiness, and depressed mood will usually be diagnosed with early Alzheimer's disease or some variant of cognitive decline. A 35-year-old presenting with bilateral leg numbness, fatigue, and visual disturbance will be worked up for multiple sclerosis. A 50-year-old presenting with peripheral burning and tingling, often with concurrent metabolic syndrome, will be told they have diabetic neuropathy. A 25-year-old presenting with fatigue, depressed mood, and difficulty concentrating will be diagnosed with major depressive disorder.
In each of those scenarios, a meaningful subset of patients turns out to have B12 deficiency as the actual cause — sometimes alongside the suspected condition, sometimes instead of it. The masquerade is so thorough that:
- Multiple sclerosis — severe B12 deficiency can produce white matter lesions on brain MRI that look nearly indistinguishable from MS lesions. Patients have been started on disease-modifying MS therapy when the underlying problem was B12 deficiency.
- Dementia and Alzheimer's disease — "pseudodementia" from B12 deficiency can be completely reversible with repletion. The American Academy of Neurology recommends B12 testing in every dementia workup.
- Depression — low B12 is significantly more common in depressed patients than in the general population, and B12 deficiency can produce depression as a primary symptom through SAMe-dependent serotonin synthesis pathways.
- Diabetic neuropathy — metformin-treated diabetics have a 10-30% prevalence of B12 deficiency. The neuropathy that's blamed on the diabetes is often partly or wholly driven by B12 deficiency that the metformin caused.
- Chronic fatigue and ME/CFS — the profound, unrelenting fatigue of B12 deficiency overlaps closely with the chronic fatigue syndromes. High-dose hydroxocobalamin injections produce dramatic improvement in some ME/CFS patients.
- Postpartum depression — pregnancy and lactation deplete maternal B12 stores significantly. Postpartum B12 deficiency contributes to postpartum mood disorders.
- Parkinson's disease, restless legs syndrome, and small-fiber neuropathy — all have documented associations with marginal B12 status.
The cost of missing B12 deficiency is high (sometimes irreversible neurological damage), and the cost of testing for it is trivial. Serum B12, MMA, and homocysteine together cost less than $100 in most US laboratories and should be ordered any time the differential includes a presentation that B12 deficiency could explain.
MMA as the Definitive Marker
Serum B12 is a poor test. Its reference range (typically 200-900 pg/mL) is widely acknowledged to be too broad on the low end — patients with serum B12 levels of 250-450 pg/mL who would be called "normal" by the lab routinely have clinical B12 deficiency that responds to repletion. The reasons:
- Serum B12 includes both biologically active B12 (bound to transcobalamin II) and inactive haptocorrin-bound B12 that cannot be delivered to cells. The active fraction is typically only 10-30% of the total.
- Many assays cross-react with inactive B12 analogues, especially in patients consuming spirulina or fermented foods.
- Serum B12 drops only after liver stores are substantially depleted — by which point neurological damage may already be progressing.
Methylmalonic acid (MMA) is the most sensitive and most specific functional marker of B12 deficiency available. The biology is straightforward: B12 (as adenosylcobalamin) is the cofactor for methylmalonyl-CoA mutase, which converts methylmalonyl-CoA to succinyl-CoA. When B12 is insufficient at the tissue level, this reaction slows, methylmalonyl-CoA accumulates, and excess methylmalonic acid spills into the blood and urine. MMA elevation therefore reflects functional B12 deficiency at the cellular level, not just serum levels.
MMA rises before serum B12 falls. It rises in patients with "normal" serum B12 who have clinical neurological symptoms. It distinguishes true B12 deficiency from elevated homocysteine due to folate deficiency or renal dysfunction. The combination of serum B12 + MMA + homocysteine is the modern diagnostic workup. Holotranscobalamin (holoTC) — the active fraction of B12 actually bound to its tissue-delivery protein — is the earliest marker and may be the future standard, but it is not yet widely available in the US.
Practical interpretation:
- Serum B12 below 200 pg/mL with elevated MMA and homocysteine = clear B12 deficiency, treat immediately
- Serum B12 200-400 pg/mL with elevated MMA = functional B12 deficiency, treat (this is the group most often missed)
- Serum B12 400-900 pg/mL with normal MMA and homocysteine = adequate
- Serum B12 below 1000 pg/mL with clinical symptoms suggesting B12 deficiency = trial of repletion is reasonable regardless of MMA
Methylcobalamin vs Cyanocobalamin in Neurological Dosing
The form of B12 used for repletion matters most in patients with neurological involvement. There are four forms in clinical use:
- Cyanocobalamin — synthetic, cheapest, most stable. Contains a cyanide group that the body must remove and the molecule converted to active forms. Effective at raising serum B12 but requires hepatic processing.
- Hydroxocobalamin — naturally occurring, longest tissue retention, preferred for IM injection in Europe. Strong nitric-oxide scavenger. The form used as the cyanide poisoning antidote (Cyanokit).
- Methylcobalamin — the methylated coenzyme form active in the methionine synthase reaction. Crosses cell membranes and is incorporated directly into the methylation cycle. Preferred form for neurological indications.
- Adenosylcobalamin (dibencozide) — the coenzyme form active in the methylmalonyl-CoA mutase reaction in mitochondria. Less commonly available; often combined with methylcobalamin.
For patients with peripheral neuropathy, subacute combined degeneration, or other neurological B12 deficiency, integrative neurologists and many conventional neurology consultants prefer methylcobalamin or hydroxocobalamin over cyanocobalamin. The reasoning:
- Methylcobalamin is active in the methionine synthase reaction without requiring metabolic conversion. In the brain and peripheral nerves, where local B12 conversion may be impaired, this matters.
- Cyanocobalamin requires removal of the cyanide moiety. In patients with smoking-related chronic low-grade cyanide exposure (cigarette smokers), or with renal impairment that slows cyanide clearance, this can become a meaningful issue.
- Hydroxocobalamin has the longest tissue retention — a single IM dose persists for weeks.
- Methylcobalamin sublingual tablets are inexpensive, well-tolerated, and bypass the entire gastrointestinal absorption cascade (relevant in pernicious anemia, post-bariatric surgery, atrophic gastritis, and any malabsorption).
Cyanocobalamin still works — it has been the workhorse of B12 therapy for decades and most patients respond well. But for neurological B12 deficiency, methylcobalamin and hydroxocobalamin are preferred in the integrative and natural medicine literature.
High-Dose 5000 mcg Sublingual Protocol
For patients with peripheral neuropathy or other neurological B12 deficiency who cannot or prefer not to receive IM injections, high-dose sublingual methylcobalamin is the alternative. The dose used clinically is 5000 mcg (5 mg) once daily, dissolved under the tongue. This is approximately 2000 times the official daily requirement (2.4 mcg), but the dose is necessary because:
- Sublingual absorption is variable and incomplete — only a fraction of the dose reaches the bloodstream
- The therapeutic goal is to drive tissue concentrations high enough to saturate methionine synthase activity, not just to meet basal needs
- Damaged or demyelinating nerves have higher local B12 demand than healthy nerves
- Excess B12 is harmless — the kidney filters and excretes whatever isn't taken up, and no upper tolerable limit has ever been established
The 5000 mcg sublingual protocol parallels the standard 1000 mcg IM injection protocol but allows the patient to self-administer at home daily. The Japanese neurological literature has used high-dose methylcobalamin (up to 6 mg/day oral) for diabetic neuropathy, Bell's palsy, trigeminal neuralgia, and other peripheral nerve conditions since the 1970s with consistent positive findings.
Practical implementation:
- Initial 4 weeks: 5000 mcg methylcobalamin sublingually each morning, on an empty stomach, allowed to dissolve fully under the tongue (not swallowed). Add hydroxocobalamin 1000 mcg IM weekly during this phase if available.
- Weeks 4-12: Continue 5000 mcg sublingual daily; reassess symptoms; re-check serum B12 (should be supraphysiological at this point, >1500 pg/mL), MMA (should be normal), and homocysteine (should be normal).
- Maintenance (month 3+): Reduce to 1000-2000 mcg sublingual daily, indefinitely. Patients with pernicious anemia or other malabsorption causes need lifelong supplementation; vegans and PPI users need it for as long as the underlying cause persists.
For severe SCD or rapidly progressive symptoms, parenteral hydroxocobalamin or methylcobalamin is preferred over sublingual — the daily IM injection protocol (1000 mcg/day for 1-2 weeks, then weekly for 4-8 weeks, then monthly indefinitely) reliably achieves maximum tissue saturation.
Recovery Timeline and What to Expect
Recovery from B12-related neurological damage follows a predictable timeline, with the caveat that structural axonal damage does not reverse — recovery reflects remyelination of surviving axons plus restoration of conduction.
- Days 1-7: Subjective sense of increased energy. Hematological response begins (reticulocytosis, falling MCV).
- Weeks 1-4: Burning, tingling, and paresthesias often worsen temporarily as nerves re-awaken — this is a positive sign of returning nerve function, not a sign of treatment failure. Hemoglobin rises; MCV begins to normalize.
- Months 1-3: Sensory symptoms (numbness, tingling, burning) begin to improve in a measurable way. Balance and gait may show subtle improvement.
- Months 3-6: Steady remyelination phase. Motor strength improves if it was affected. Cognitive symptoms (concentration, memory) improve if they were affected. Mood improves.
- Months 6-12: Continued improvement, particularly in proprioception and balance. Maximum recovery is typically achieved by 12-18 months. Any deficits remaining at 18-24 months are likely to be permanent.
Predictors of better recovery: shorter duration of deficiency before treatment, less severe baseline symptoms, presence of preserved sensation (axon survival), normal MRI of the spinal cord, and rapid normalization of MMA on treatment. Predictors of incomplete recovery: long duration of untreated deficiency, severe baseline neurological deficits, spinal cord MRI showing T2 hyperintensities in the dorsal columns (consistent with axonal loss), and slow MMA response.
Combination Strategies
- B12 + Folate (L-methylfolate, 1-15 mg/day) — the methionine synthase reaction requires both B12 and 5-MTHF as substrates. Folate alone can correct the anemia of B12 deficiency through the methyl trap but does not fix the neurological problem; B12 alone may be limited if folate is also depleted. Always co-supplement.
- B12 + Vitamin B6 (P5P, 25-100 mg/day) — B6 clears homocysteine via the transsulfuration pathway. The B6/B9/B12 "homocysteine triad" is the standard combination for neurological B12 deficiency.
- B12 + Alpha Lipoic Acid (600 mg/day) — ALA addresses the mitochondrial and microvascular components of peripheral neuropathy while B12 addresses the myelin component. The combination is the most evidence-based natural medicine stack for diabetic peripheral neuropathy.
- B12 + Benfotiamine (300 mg/day) — the fat-soluble form of thiamine. Pairs naturally with ALA and B12 for diabetic neuropathy support.
- B12 + Acetyl-L-Carnitine (1500-3000 mg/day) — ALCAR supports mitochondrial function in peripheral nerves and has independent evidence for diabetic neuropathy. Combination is logical.
- B12 + Magnesium glycinate (300-400 mg/day) — magnesium supports nerve excitability and may reduce the burning quality of neuropathic pain.
- B12 + omega-3 EPA/DHA (2-3 g/day) — omega-3 fatty acids are membrane components of neuronal myelin and support nerve regeneration.
Cautions
- Acute hypokalemia during rapid repletion — severe pernicious anemia patients beginning B12 therapy can develop dangerously low potassium as new red blood cells are produced and pull potassium into the intracellular compartment. Check potassium during the first week of treatment and supplement as needed.
- Initial worsening of paresthesias — common, transient, and a positive sign of returning nerve function. Warn patients in advance.
- Folic acid masking B12 deficiency — if folate is supplemented without B12, the anemia corrects but the neurological damage progresses silently. Always check B12 status before initiating high-dose folate.
- Cyanocobalamin in smokers and renal impairment — chronic cigarette smoking exposes the body to low-grade cyanide. Renal impairment slows cyanide clearance. In both situations, methylcobalamin or hydroxocobalamin is preferable to cyanocobalamin.
- Acne and rosacea flares — high-dose B12 (especially cyanocobalamin) can occasionally trigger acne breakouts or rosacea flares. Switching to methylcobalamin or hydroxocobalamin often resolves the issue.
- Nitrous oxide avoidance — recreational use of nitrous oxide ("whippets") irreversibly inactivates B12. Patients with marginal B12 status can develop acute, severe myeloneuropathy after even a single heavy exposure. Avoid recreational N2O entirely if B12 status is borderline; ensure aggressive B12 repletion before elective procedures involving N2O anesthesia.
Key Research Papers
- Lindenbaum J et al. (1988). Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. NEJM. — PubMed
- Healton EB et al. (1991). Neurologic aspects of cobalamin deficiency. Medicine (Baltimore). — PubMed
- Stabler SP (2013). Vitamin B12 deficiency. NEJM. — PubMed
- Andres E et al. (2004). Vitamin B12 (cobalamin) deficiency in elderly patients. CMAJ. — PubMed
- Kuwabara S et al. (1999). Intravenous methylcobalamin treatment for uremic and diabetic neuropathy. Internal Medicine. — PubMed
- Sun Y et al. (2005). Effectiveness of vitamin B12 on diabetic neuropathy: systematic review of clinical controlled trials. Acta Neurologica Taiwanica. — PubMed
- Yaqub BA et al. (1992). Subacute combined degeneration of the spinal cord. Brain. — PubMed
- Garakani A et al. (2014). Vitamin B12 deficiency and depression: a systematic review. Indian J Psychiatry. — PubMed
- Carmel R (2008). How I treat cobalamin (vitamin B12) deficiency. Blood. — PubMed
- Garcia-Cazorla A et al. (2008). Genetic disorders of cobalamin transport and metabolism. Pediatric Neurology. — PubMed
- Briani C et al. (2013). Cobalamin deficiency: clinical picture and radiological findings. Nutrients. — PubMed
- Hathout L, El-Saden S (2011). Nitrous oxide-induced B12 deficiency myelopathy. J Neurol Sci. — PubMed
PubMed Topic Searches
- PubMed: vitamin B12 peripheral neuropathy
- PubMed: methylcobalamin neurological disease
- PubMed: subacute combined degeneration B12
- PubMed: MMA functional B12
- PubMed: B12 great masquerader
Connections
- Vitamin B12’s Impossible Journey — interactive animation
- Vitamin B12 Overview
- B12 Benefits Hub
- B12 for Anemia & Hematology
- B12 for Cognition & Methylation
- B12 for Vegans & Absorption
- B12 and the Nervous System
- B12 Deficiency Diagnosis
- Peripheral Neuropathy
- ALA for Diabetic Neuropathy
- Diabetes
- Numbness & Tingling
- Vitamin B9 (Folate)
- Vitamin B6
- Vitamin B1 (Thiamine / Benfotiamine)
- Homocysteine
- Vitamin B12 Test
- Methionine
- Chronic Pain