Vitamin B12 for Cognition and Methylation
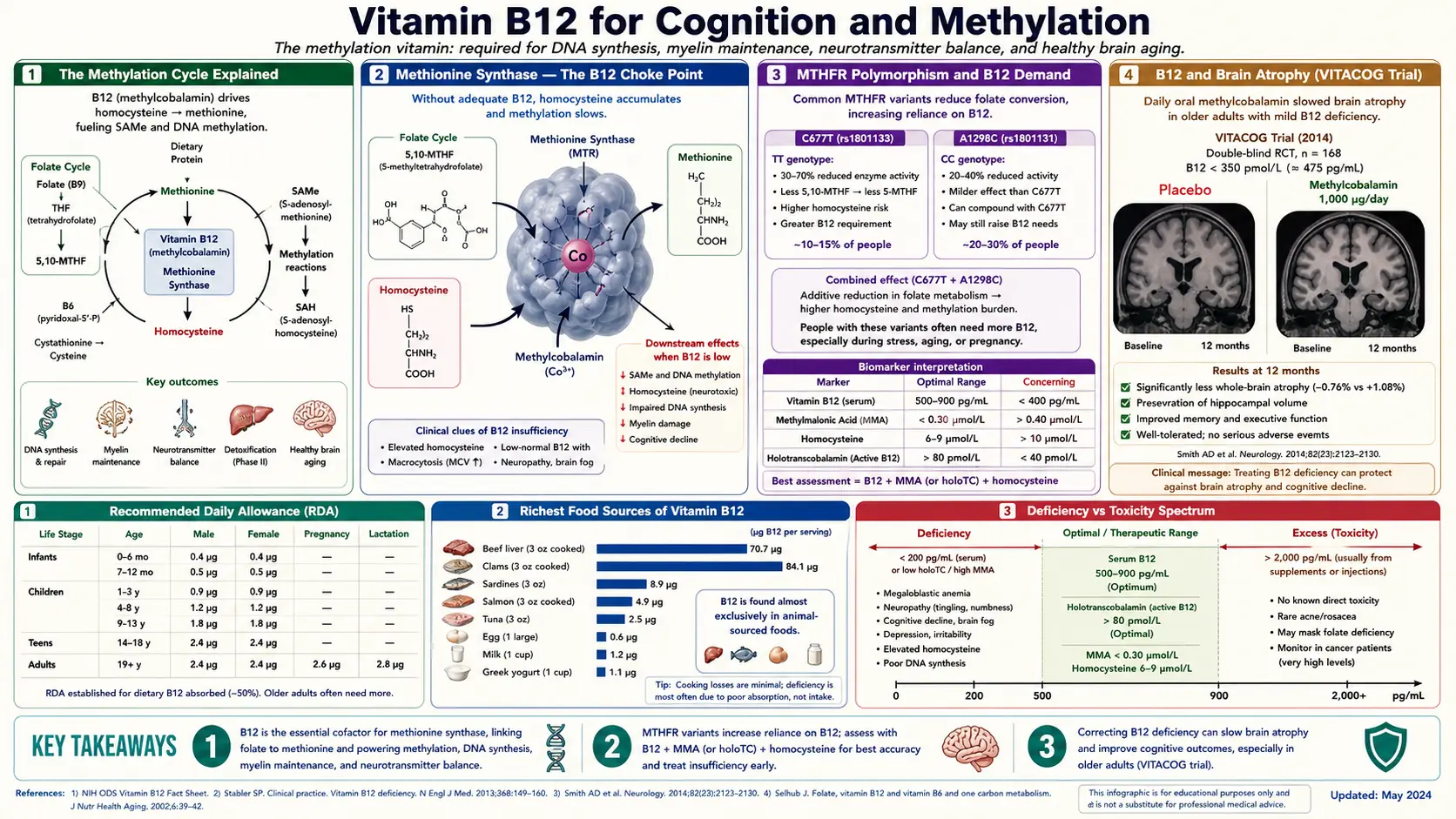
The methylation cycle is the most consequential biochemical reaction sequence in the human body that most people have never heard of. Methyl groups are added to and removed from DNA, RNA, proteins, phospholipids, and small molecules thousands of times per second in every cell, controlling gene expression, neurotransmitter synthesis, myelin maintenance, hormone metabolism, and homocysteine clearance. The whole system pivots on a single B12-dependent enzyme: methionine synthase. Low B12 collapses methylation, drives homocysteine upward, and accelerates brain atrophy. This deep-dive walks through the methylation cycle in clinical detail, the MTHFR polymorphism that increases B12 demand, the 10-20% prevalence of low B12 in elderly cognitive decline, the SAMe and monoamine-synthesis mechanism behind B12's antidepressant effect, and the VITACOG trial showing 53% reduction in brain atrophy with B-vitamin supplementation.
Interactive Visualization Vitamin B12’s Impossible Journey Five stages, two carrier proteins, a working stomach and one specific stretch of gut. Break any link — a PPI, metformin, pernicious anaemia — and absorption collapses. Launch →
Table of Contents
- The Methylation Cycle Explained
- Methionine Synthase — The B12 Choke Point
- MTHFR Polymorphism and B12 Demand
- B12 and Brain Atrophy (VITACOG Trial)
- B12 and Dementia — The Elderly Connection
- Depression and SAMe Mechanism
- Monoamine Synthesis Dependency
- B-Complex Protocols for Memory and Mood
- Cautions
- Key Research Papers
- Connections
- Featured Videos
The Methylation Cycle Explained
The methylation cycle is the set of reactions that produces and recycles the cellular methyl-group currency that drives more than 200 distinct biochemical reactions. The center of the cycle is the conversion of methionine to S-adenosylmethionine (SAMe) — the universal methyl donor. When SAMe gives up its methyl group (to DNA, to a neurotransmitter precursor, to a phospholipid, etc.), it becomes S-adenosylhomocysteine (SAH), which is then hydrolyzed to homocysteine.
Homocysteine has three possible fates:
- Remethylation to methionine via methionine synthase (requires B12 as cofactor and 5-MTHF as methyl donor) — the dominant pathway in most tissues
- Remethylation to methionine via betaine-homocysteine methyltransferase (BHMT) (requires betaine/TMG as methyl donor) — a secondary pathway predominantly in liver and kidney
- Transsulfuration to cysteine via cystathionine beta-synthase (CBS) (requires B6 as cofactor) — the irreversible disposal pathway that feeds glutathione synthesis
The relative flux through these pathways depends on the body's current needs. When SAMe is abundant, it allosterically activates CBS and inhibits BHMT, pushing homocysteine into the transsulfuration pathway for disposal. When SAMe is depleted, the brakes come off remethylation and homocysteine cycles back to methionine and SAMe to restore the pool.
Methylation downstream of SAMe touches:
- DNA methylation — the epigenetic marking of gene promoter regions that controls which genes are turned on or off. Affects every cell, every day. Aberrant DNA methylation is implicated in cancer, aging, neurodegeneration, and developmental disorders.
- Histone methylation — chemical modifications of the proteins that package DNA into chromatin, controlling gene accessibility.
- Neurotransmitter synthesis — methylation steps in the production and degradation of dopamine, norepinephrine, epinephrine, serotonin, and melatonin. Catechol-O-methyltransferase (COMT) and phenylethanolamine N-methyltransferase (PNMT) are SAMe-dependent.
- Phospholipid synthesis — conversion of phosphatidylethanolamine to phosphatidylcholine via phosphatidylethanolamine N-methyltransferase (PEMT). Phosphatidylcholine is the major lipid component of cell membranes and a critical building block of myelin.
- Creatine synthesis — guanidinoacetate methyltransferase (GAMT) uses SAMe to make creatine. Creatine biosynthesis consumes ~75% of the body's daily SAMe production.
- Hormone metabolism — estrogen, thyroid hormone, and other steroid hormones undergo methylation reactions in their metabolism and detoxification.
- Carnitine, melatonin, and many other small molecules — SAMe-dependent synthesis steps.
When methylation collapses (most commonly because of B12 deficiency, less commonly because of folate deficiency, MTHFR polymorphism, B2/B6 deficiency, or BHMT/CBS variants), the downstream effects radiate across every cell type. The brain is particularly vulnerable because of its high methylation demand: neurotransmitter synthesis, myelin maintenance, and gene regulation in neurons are all methylation-dependent.
Methionine Synthase — The B12 Choke Point
Methionine synthase is the only enzyme in human biology that uses methylcobalamin as its cofactor. The reaction it catalyzes is conceptually simple but biologically critical: it transfers a methyl group from 5-methyltetrahydrofolate (5-MTHF) onto homocysteine, producing methionine and regenerating tetrahydrofolate (THF).
Two failure modes simultaneously emerge from methionine synthase inactivity:
- Homocysteine accumulates — because the major pathway for its disposal is blocked. Elevated homocysteine is an independent risk factor for cardiovascular disease, stroke, cognitive decline, dementia, neural tube defects, and recurrent miscarriage.
- Folate gets stuck — the so-called "methyl trap." 5-MTHF is normally an intermediate that quickly donates its methyl group and recycles. When methionine synthase is inactive, 5-MTHF builds up because there is nowhere for it to go (the methyl-group transfer is the only enzymatic exit from 5-MTHF). Meanwhile, THF (the form needed for thymidine synthesis and purine synthesis) is not regenerated. The cell becomes functionally folate-deficient even when total folate is adequate.
The methyl trap is the molecular explanation for why B12 deficiency produces an anemia that looks exactly like folate deficiency — the underlying defect is the same (insufficient active folate for DNA synthesis), it's just reached via different upstream causes.
Methionine synthase activity also depends on the cell's redox state. The cobalt at the heart of B12 must be maintained in the reduced (Co+1) state to accept and transfer the methyl group. Oxidative stress periodically oxidizes the cobalt to Co+2, inactivating the enzyme. The cell uses an auxiliary enzyme called methionine synthase reductase (encoded by MTRR) to reactivate it. MTRR polymorphisms are increasingly recognized as a factor in functional B12 deficiency — some people with adequate B12 still have low methionine synthase activity because their MTRR is less efficient.
MTHFR Polymorphism and B12 Demand
MTHFR (methylenetetrahydrofolate reductase) is the enzyme that produces 5-MTHF — the folate form that methionine synthase needs. Two common polymorphisms reduce MTHFR activity:
- C677T (rs1801133) — the more clinically important variant. Heterozygotes (CT) have ~35% reduced enzyme activity; homozygotes (TT) have ~70% reduced activity. Approximately 10-15% of people of European descent are TT homozygous; rates are higher in some Hispanic and lower in some African populations.
- A1298C (rs1801131) — less impact on enzyme activity but can compound C677T effects when both variants coexist (compound heterozygosity).
The clinical consequences of reduced MTHFR activity include:
- Higher homocysteine levels at any given folate and B12 intake
- Reduced 5-MTHF generation, which can functionally starve methionine synthase even when B12 is adequate
- Increased risk for neural tube defects in offspring of TT mothers
- Association with cardiovascular disease in some populations
- Association with depression, particularly treatment-resistant depression
- Possible association with some cancers (the literature is mixed)
The clinical implication for B12: patients with MTHFR variants need more of the upstream cofactors (B12, B2, B6) and benefit from supplementation with the already-methylated folate form (L-5-methyltetrahydrofolate, often abbreviated as L-methylfolate, LMTHF, or just "methylfolate") rather than folic acid. L-methylfolate bypasses the MTHFR step entirely — the body doesn't need to reduce it before use. This approach (combined methylcobalamin + L-methylfolate, plus B6 as P5P and B2 as R5P) is the standard "methylated B-complex" protocol used in integrative psychiatry for treatment-resistant depression with MTHFR variants.
Cautions:
- MTHFR variants are common (one-third of the population has at least one C677T allele); their presence does not mean a person needs lifelong special supplementation unless they have clinical or biochemical evidence of dysfunction (elevated homocysteine, low folate or B12, depression, etc.). The variant alone is not a disease.
- Methylfolate at very high doses (15+ mg/day) can occasionally cause overstimulation, anxiety, or insomnia — especially in patients with COMT variants or histamine-sensitivity profiles. Start low (400-800 mcg) and titrate upward.
B12 and Brain Atrophy (VITACOG Trial)
The VITACOG trial (Smith et al., 2010, PLoS ONE) is the landmark study demonstrating that B vitamins can slow brain atrophy in mild cognitive impairment. The trial randomized 271 elderly patients with mild cognitive impairment (MCI — the prodromal state preceding Alzheimer's disease) to one of two arms for 2 years:
- Active arm: folic acid 0.8 mg + cyanocobalamin 0.5 mg + vitamin B6 (pyridoxine) 20 mg daily
- Placebo arm: identical-appearing placebo tablets
The primary outcome was the change in whole-brain volume measured by MRI at baseline and at 2 years. Brain atrophy in healthy elderly people occurs at a rate of approximately 0.5% per year; in MCI patients it accelerates to 1-1.5% per year; in Alzheimer's disease it approaches 2.5% per year.
Results:
- Mean rate of brain atrophy in the B-vitamin arm: 0.76%/year
- Mean rate of brain atrophy in the placebo arm: 1.08%/year
- Relative reduction in atrophy: 29.6% overall
- In the subgroup with baseline homocysteine above the median (>11 micromol/L), the reduction was even more dramatic: 53% reduction in atrophy rate
- The effect was driven entirely by patients with elevated homocysteine — in those with low baseline homocysteine, B vitamins produced no benefit
A follow-up VITACOG cognitive analysis (de Jager et al., 2012) showed that the B-vitamin arm preserved cognitive function on standardized testing better than placebo, again primarily in the elevated-homocysteine subgroup. A subsequent MRI sub-study (Douaud et al., 2013) demonstrated that the B-vitamin arm specifically preserved the brain regions most vulnerable to Alzheimer's disease — the medial temporal lobes, including the hippocampus.
The VITACOG findings established three critical principles:
- B-vitamin supplementation can slow brain atrophy in at-risk elderly patients
- The benefit is driven by homocysteine reduction — not by some other mechanism
- The intervention works only in patients with elevated baseline homocysteine; patients with already-low homocysteine get no incremental benefit
Practical implication: every patient with MCI, family history of dementia, or risk factors for cognitive decline should have homocysteine measured. If homocysteine is above 10-11 micromol/L (which is most elderly patients), a B12 + folate + B6 trial is reasonable. Target homocysteine reduction to below 9 micromol/L.
B12 and Dementia — The Elderly Connection
Low B12 is found in approximately 10-20% of elderly patients presenting with cognitive decline. The mechanisms are multiple:
- Atrophic gastritis — affects roughly 30% of adults over 60, reducing gastric acid output and impairing protein-bound B12 extraction from food. Stomach acid is needed to free B12 from food protein; without it, B12 passes through undigested. The condition is often asymptomatic and unrecognized.
- Reduced intrinsic factor production — some elderly patients have insufficient intrinsic factor without overt pernicious anemia. Borderline IF deficiency progresses gradually and may not be picked up by anti-IF antibody testing.
- Polypharmacy — PPIs (omeprazole, esomeprazole, pantoprazole), H2 blockers (famotidine, ranitidine historically), and metformin all reduce B12 absorption. Many elderly patients are on at least one and often several of these medications for years.
- Decreased dietary intake — elderly patients often eat less meat, less variety, and may shift toward lower-cost soft foods with lower B12 content.
- Slower hepatic stores depletion — means deficiency can develop over many years without obvious clinical onset, presenting eventually as gradual cognitive decline that is easy to attribute to "normal aging."
The pseudodementia presentation: a subset of elderly patients with B12 deficiency present with cognitive symptoms severe enough to look like dementia — memory loss, confusion, executive dysfunction, sometimes psychotic features. These patients are frequently misdiagnosed with Alzheimer's disease. Aggressive B12 repletion (IM hydroxocobalamin 1000 mcg daily for 1-2 weeks, then weekly for 4-8 weeks, then monthly indefinitely) can produce dramatic cognitive recovery in pseudodementia patients — sometimes complete reversal of symptoms within 3-6 months.
The American Academy of Neurology, the Alzheimer's Association, and most dementia consensus guidelines recommend B12 (with MMA if borderline) as part of every dementia workup. The cost is trivial; the cost of missing reversible cognitive impairment is high. Even in patients who turn out to have primary Alzheimer's disease, optimizing B12 status reduces the additive contribution of B12 deficiency to their cognitive trajectory.
For patients with established Alzheimer's disease and low or borderline B12, B12 repletion does not reverse the AD pathology but may slow disease progression and provide modest cognitive benefit. The intervention is low-risk and inexpensive.
Depression and SAMe Mechanism
The link between B12 deficiency and depression is biologically grounded in the methylation cycle. Three mechanisms operate in parallel:
- SAMe deficiency — SAMe is itself an antidepressant. Multiple randomized controlled trials have shown that exogenous SAMe (typically 800-1600 mg/day) has antidepressant efficacy comparable to first-line SSRIs in major depressive disorder. When B12 deficiency reduces endogenous SAMe production, this antidepressant background is lost.
- Monoamine synthesis impairment — serotonin synthesis from tryptophan, dopamine synthesis from tyrosine, and norepinephrine synthesis all depend on tetrahydrobiopterin (BH4) as a cofactor. BH4 production and regeneration are linked to the methylation cycle; B12 deficiency indirectly reduces BH4 availability.
- Homocysteine neurotoxicity — elevated homocysteine is itself neurotoxic, activating NMDA receptors, generating oxidative stress in brain tissue, and promoting cerebrovascular damage. The depression of B12 deficiency may be partly a homocysteine-induced phenomenon.
The clinical evidence:
- Low B12 is significantly more common in depressed patients than in matched controls
- Low B12 is significantly more common in treatment-resistant depression than in treatment-responsive depression
- B12 supplementation augments antidepressant response in patients with low or borderline B12
- Folic acid (and especially L-methylfolate) augments antidepressant response in patients with low folate or MTHFR variants — the Trivedi 2012 SUN-D trial established 15 mg L-methylfolate as an effective augmentation strategy
- The combination of B12 + L-methylfolate + SAMe is used clinically as a "methylation-support" protocol for treatment-resistant depression
Cautions:
- B12 alone does not treat primary depression in patients with adequate baseline B12. The intervention is targeted at the subgroup with deficiency or borderline status.
- Severe depression should be treated by qualified mental-health practitioners. B12 optimization is an adjunct, not a replacement, for evidence-based depression treatment.
- The methyl-donor protocols (high-dose methylfolate, methylcobalamin, SAMe) can rarely trigger overstimulation, anxiety, or insomnia in sensitive individuals — start low and titrate.
Monoamine Synthesis Dependency
The synthesis of dopamine, norepinephrine, epinephrine, serotonin, and melatonin all depend on a small handful of cofactors. Methylation supports several steps:
- BH4 regeneration — tetrahydrobiopterin, the cofactor for tyrosine hydroxylase (TH, the rate-limiting enzyme in dopamine synthesis) and tryptophan hydroxylase (TPH, the rate-limiting enzyme in serotonin synthesis), depends on functional methylation cycles for proper regeneration.
- COMT and PNMT — SAMe-dependent enzymes in catecholamine metabolism. COMT degrades dopamine, norepinephrine, and epinephrine; PNMT converts norepinephrine to epinephrine.
- Melatonin synthesis — serotonin is converted to N-acetylserotonin by N-acetyltransferase, then N-acetylserotonin is converted to melatonin by hydroxyindole O-methyltransferase (HIOMT), a SAMe-dependent enzyme.
- Phosphatidylcholine — required for membrane integrity in synaptic terminals; produced by SAMe-dependent methylation of phosphatidylethanolamine.
When methylation collapses from B12 deficiency, all of these pathways become less efficient. The cumulative effect on mood, cognition, sleep, and motivation is what we recognize clinically as the psychiatric syndrome of B12 deficiency — depression, anhedonia, slowed thinking, poor concentration, sleep disturbance, and in severe cases frank psychiatric illness ("megaloblastic madness").
This is why B12 repletion in deficient patients often produces such striking improvements in mental energy, mood, and clarity within weeks: the methylation cycle resumes, neurotransmitter synthesis catches up, membrane phospholipids are repaired, and the entire central nervous system functions better.
B-Complex Protocols for Memory and Mood
The synergy among B vitamins means that single-nutrient supplementation often underperforms a coordinated B-complex approach. The standard protocols in integrative practice:
Cognitive Decline / MCI Prevention (Modeled on VITACOG)
- Methylcobalamin 1000 mcg daily (or hydroxocobalamin 1000 mcg IM monthly)
- L-methylfolate 400-1000 mcg daily (or folic acid 800 mcg if no MTHFR variants)
- Vitamin B6 (P5P) 25-50 mg daily
- Vitamin B2 (riboflavin or R5P) 25-50 mg daily — supports MTHFR enzyme function
- Target homocysteine below 9 micromol/L
Treatment-Resistant Depression Augmentation
- Methylcobalamin 1000-5000 mcg daily
- L-methylfolate 7.5-15 mg daily (the Deplin / Trivedi protocol)
- Vitamin B6 (P5P) 25-100 mg daily
- Vitamin B2 (R5P) 25 mg daily
- Consider SAMe 800-1600 mg/day in divided doses
- Consider omega-3 EPA > 1000 mg daily
- Consider vitamin D optimization (target 50-80 ng/mL)
- Continue prescribed antidepressant per psychiatrist
Cognitive Performance Enhancement (No Deficiency)
- Methylcobalamin 500-1000 mcg daily
- L-methylfolate 400-800 mcg daily
- Vitamin B6 (P5P) 10-25 mg daily
- Vitamin B2 (R5P) 10-25 mg daily
- Plus other cognitive supports per individual (e.g., omega-3, magnesium L-threonate, exercise, sleep optimization)
The protocols are well-tolerated and inexpensive. Daily methylcobalamin 1000 mcg costs roughly $5-10 per month; methylfolate is similar; B6 and B2 are cheaper still. The combined methylated B-complex monthly cost is typically under $30.
Cautions
- Methylation overstimulation — in some patients, particularly those with COMT slow variants or histamine sensitivity, high-dose methylated B-complex protocols can trigger anxiety, insomnia, irritability, or headache. Start at the low end (methylfolate 400 mcg, methylcobalamin 1000 mcg) and titrate slowly.
- Folic acid masking B12 deficiency — if folate is given without B12, the anemia corrects but the neurological problem progresses. Always check B12 status before high-dose folate.
- MTHFR variant testing — useful in patients with elevated homocysteine, treatment-resistant depression, or recurrent miscarriage. Not necessary for everyone — the polymorphism is common (one-third of the population has at least one C677T allele) and most carriers are clinically asymptomatic.
- Drug interactions — methylfolate can theoretically interact with methotrexate (a folate antagonist used in rheumatology and oncology); discuss with the prescribing physician. SAMe should be avoided in patients with bipolar disorder (can precipitate mania).
- Bipolar disorder caution — aggressive methylation support, especially SAMe, can trigger manic or hypomanic episodes in bipolar patients. Use cautiously and only with psychiatric oversight.
- Don't replace prescribed psychiatric treatment — the B-complex protocols are adjuncts to evidence-based psychiatric care, not substitutes for it. Severe depression and dementia require professional management.
Key Research Papers
- Smith AD et al. (2010). Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: the VITACOG randomised controlled trial. PLoS ONE. — PubMed
- de Jager CA et al. (2012). Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: a randomized controlled trial. Int J Geriatr Psychiatry. — PubMed
- Douaud G et al. (2013). Preventing Alzheimer's disease-related gray matter atrophy by B-vitamin treatment. PNAS. — PubMed
- Clarke R et al. (1998). Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol. — PubMed
- Seshadri S et al. (2002). Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. NEJM. — PubMed
- Trivedi MH et al. (2012). L-methylfolate as adjunctive therapy for SSRI-resistant major depression. Am J Psychiatry. — PubMed
- Coppen A, Bailey J (2000). Enhancement of the antidepressant action of fluoxetine by folic acid. J Affect Disord. — PubMed
- Papakostas GI et al. (2010). S-adenosyl methionine (SAMe) augmentation of serotonin reuptake inhibitors for antidepressant nonresponders. Am J Psychiatry. — PubMed
- Bottiglieri T (2002). S-Adenosyl-L-methionine (SAMe): from the bench to the bedside — molecular basis of a pleiotrophic molecule. Am J Clin Nutr. — PubMed
- Refsum H et al. (2006). The Hordaland Homocysteine Study: a community-based study of homocysteine, its determinants, and associations with disease. J Nutr. — PubMed
- Frosst P et al. (1995). A candidate genetic risk factor for vascular disease: a common mutation in MTHFR. Nature Genetics. — PubMed
- Selhub J et al. (1995). Vitamin status and intake as primary determinants of homocysteinemia in an elderly population. JAMA. — PubMed
PubMed Topic Searches
- PubMed: B12 folate B6 homocysteine brain atrophy
- PubMed: methylation cycle depression treatment-resistant
- PubMed: MTHFR polymorphism depression methylfolate
- PubMed: B12 dementia cognitive decline elderly
- PubMed: SAMe depression augmentation
Connections
- Vitamin B12’s Impossible Journey — interactive animation
- Vitamin B12 Overview
- B12 Benefits Hub
- B12 for Nerve Health
- B12 for Anemia
- B12 for Vegans & Absorption
- B12 and the Nervous System
- Vitamin B9 (Folate / Methylfolate)
- Vitamin B6
- Vitamin B2 (Riboflavin)
- Homocysteine
- Vitamin B12 Test
- Depression
- Dementia
- Alzheimer's Disease
- Methionine
- Brain Fog
- Fatigue