Vitamin B12 and Nervous System Health
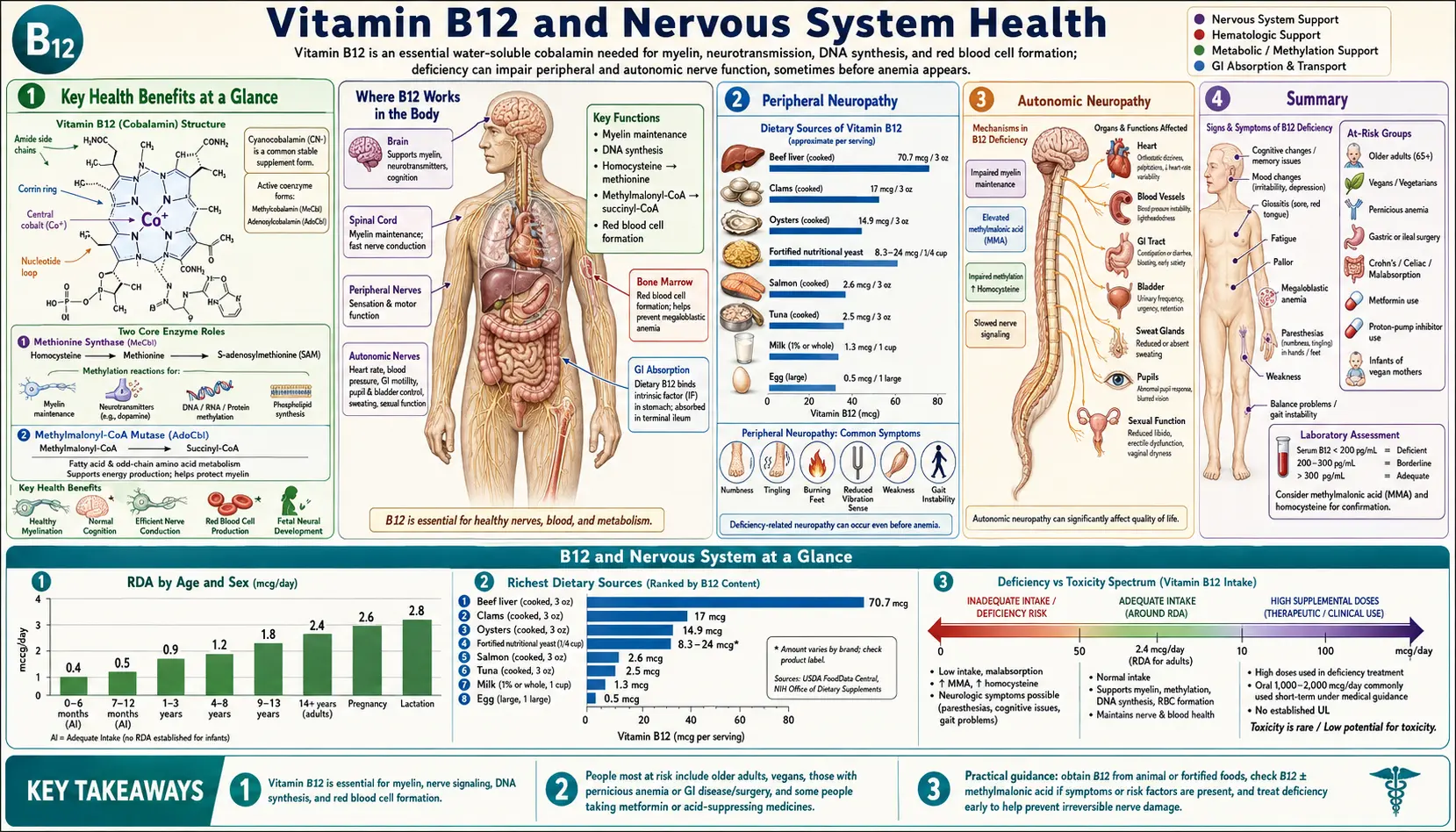
Vitamin B12 (cobalamin) occupies a uniquely critical position in nervous system health. Unlike most other vitamins, B12 deficiency can produce irreversible neurological damage if not recognized and treated promptly. The neurological manifestations span the full breadth of the nervous system — from peripheral nerves to the spinal cord, brain, and optic pathways — and can precede hematological abnormalities by months to years. This page summarizes the biochemistry by which B12 supports myelin synthesis and methylation, the classic and under-recognized clinical syndromes of deficiency, the imaging findings, and the treatment strategies that preserve neurological function, along with the landmark research papers that underpin modern practice.
Interactive Visualization Vitamin B12’s Impossible Journey Five stages, two carrier proteins, a working stomach and one specific stretch of gut. Break any link — a PPI, metformin, pernicious anaemia — and absorption collapses. Launch →Table of Contents
- Key Health Benefits at a Glance
- Myelin Sheath Synthesis
- S-Adenosylmethionine (SAMe) and Methylation
- Subacute Combined Degeneration of the Spinal Cord
- Peripheral Neuropathy
- Cognitive Impairment and Dementia
- Optic Neuropathy
- Autonomic Neuropathy
- B12 and Multiple Sclerosis Research
- Methylcobalamin vs. Cyanocobalamin
- Clinical Recognition and Treatment
- Summary
- Research Papers and References
- Connections
- Featured Videos
Key Health Benefits at a Glance
Before diving into the mechanism-level detail, the following is a high-level summary of the neurological consequences of adequate (and inadequate) B12 status. Supporting studies are linked in the Research Papers section.
- Preserves myelin integrity – B12 is required for methionine synthase and methylmalonyl-CoA mutase, two enzymes central to the phospholipid methylation and fatty-acid metabolism that maintain the myelin sheath.
- Prevents subacute combined degeneration – Early replacement halts and can reverse the demyelination of the dorsal columns and lateral corticospinal tracts characteristic of severe deficiency.
- Reduces peripheral neuropathy – B12 replacement improves sensory symptoms and nerve conduction in documented-deficient patients, with the best outcomes when started within 3 months of symptom onset.
- Supports cognition and lowers homocysteine – Adequate B12 keeps homocysteine in check, a vascular and neurotoxic risk factor linked to cognitive decline and dementia.
- Maintains SAMe-dependent methylation – B12 regenerates methionine and sustains SAMe, the universal methyl donor required for neurotransmitter synthesis, DNA methylation, and phospholipid maintenance in neurons.
- Protects visual pathways – Adequate B12 prevents the centrocecal-scotoma optic neuropathy sometimes seen in chronic deficiency.
- Supports autonomic function – Repletion can improve neurogenic bladder, orthostatic hypotension, and other autonomic manifestations in deficient patients.
- Prevents folate-masking catastrophe – Recognizing B12 status before folate therapy prevents the partial correction of anemia while neurological damage silently progresses.
- Evidence-based treatment protocols – Parenteral cyanocobalamin or hydroxocobalamin regimens are well established and inexpensive, with robust recovery data when initiated early.
Myelin Sheath Synthesis
The myelin sheath, the lipid-rich membrane that insulates nerve fibers and enables rapid saltatory conduction of electrical impulses, is the primary target of B12 deficiency in the nervous system.
- Myelin composition: Myelin is composed of approximately 70% lipid and 30% protein, with a uniquely high proportion of galactocerebroside, cholesterol, and phospholipids including phosphatidylcholine and phosphatidylethanolamine. The synthesis of these lipid components requires properly functioning methylation pathways, for which B12 is an essential cofactor.
- Methylmalonyl-CoA mutase pathway: B12 in the form of adenosylcobalamin serves as a cofactor for methylmalonyl-CoA mutase, which converts methylmalonyl-CoA to succinyl-CoA. When B12 is deficient, methylmalonyl-CoA accumulates and is incorporated into fatty acid synthesis in place of malonyl-CoA, producing abnormal odd-chain and branched-chain fatty acids. These abnormal fatty acids are incorporated into myelin lipids, destabilizing the myelin membrane and leading to demyelination.
- Phospholipid methylation: B12, through its role in SAMe production via the methionine synthase reaction, supports the methylation of phosphatidylethanolamine to phosphatidylcholine by the enzyme phosphatidylethanolamine N-methyltransferase. Impaired phosphatidylcholine synthesis compromises myelin membrane fluidity and structural integrity.
- Oligodendrocyte function: In the central nervous system, oligodendrocytes are responsible for myelin production and maintenance. These cells have high metabolic demands and are particularly vulnerable to B12 deficiency-induced metabolic dysfunction, contributing to the central demyelination seen in subacute combined degeneration.
S-Adenosylmethionine (SAMe) and Methylation
The methylation pathway linking B12 to nervous system function is perhaps the most critical biochemical connection in clinical neurology.
- Methionine synthase reaction: Methylcobalamin serves as a cofactor for methionine synthase, which transfers a methyl group from 5-methyltetrahydrofolate to homocysteine, regenerating methionine. Methionine is then activated to SAMe, the universal methyl donor required for over 200 methyltransferase reactions, many of which are essential for nervous system function.
- Methylation in the CNS: In the central nervous system, SAMe-dependent methylation is required for myelin basic protein and myelin-associated glycoprotein production, neurotransmitter synthesis and degradation (serotonin, dopamine, norepinephrine, melatonin), DNA methylation and epigenetic regulation in neurons, histone methylation affecting gene expression, and phospholipid methylation for neuronal membrane maintenance.
- SAMe depletion cascade: B12 deficiency leads to methionine synthase dysfunction, homocysteine accumulation, and critically, depletion of SAMe. The resulting global hypomethylation state in the nervous system affects multiple processes simultaneously, explaining the diverse neurological manifestations of B12 deficiency.
- Folate trap mechanism: B12 deficiency traps folate in the 5-methyltetrahydrofolate form, preventing its recycling back to tetrahydrofolate. This “methyl trap” simultaneously impairs folate-dependent reactions (thymidine synthesis for DNA, purine synthesis) and SAMe production, creating a dual metabolic insult in the nervous system.
Subacute Combined Degeneration of the Spinal Cord
Subacute combined degeneration (SCD) is the classic neurological syndrome of B12 deficiency and one of the few reversible causes of myelopathy if recognized early.
- Pathology: SCD is characterized by demyelination and vacuolar degeneration that preferentially affects the dorsal (posterior) columns and lateral corticospinal tracts of the spinal cord. The term “combined” refers to the simultaneous involvement of both sensory (dorsal column) and motor (corticospinal) pathways. The thoracic spinal cord is typically affected first and most severely.
- Clinical presentation: The earliest symptoms are usually symmetric paresthesias (numbness and tingling) in the hands and feet, often described as a “glove and stocking” distribution. This is followed by progressive sensory ataxia due to dorsal column dysfunction, manifesting as difficulty walking in the dark, a positive Romberg sign, and impaired proprioception and vibration sense. Spastic paraparesis develops as the lateral corticospinal tracts become involved.
- Lhermitte sign: Patients with SCD may experience Lhermitte sign, an electric shock-like sensation radiating down the spine and into the limbs upon neck flexion. While classically associated with multiple sclerosis, Lhermitte sign in the context of B12 deficiency strongly suggests spinal cord involvement.
- MRI findings: Spinal cord MRI in SCD typically demonstrates T2-hyperintense signal in the dorsal columns, often described as an inverted “V” pattern on axial imaging. These signal changes may extend over multiple spinal segments. With treatment, MRI abnormalities can resolve, though this may lag behind clinical improvement.
- Reversibility: Early SCD is largely reversible with prompt B12 replacement therapy. However, if treatment is delayed beyond 6 to 12 months of symptom onset, neurological damage may become permanent. The extent of recovery correlates inversely with the duration and severity of symptoms before treatment initiation.
Peripheral Neuropathy
Peripheral neuropathy is the most common neurological manifestation of B12 deficiency and often precedes myelopathy.
- Pattern of involvement: B12 deficiency typically produces a symmetric, length-dependent sensory polyneuropathy affecting the distal lower extremities before the upper extremities. Sensory symptoms predominate over motor involvement in most cases, though motor neuropathy can occur in severe or prolonged deficiency.
- Sensory modalities affected: Both large fiber (vibration, proprioception) and small fiber (pain, temperature) sensory modalities can be affected, though large fiber dysfunction tends to be more prominent. Loss of vibration sense at the ankles and impaired joint position sense in the toes are often the earliest objective findings on examination.
- Electrodiagnostic findings: Nerve conduction studies in B12 deficiency neuropathy typically show reduced sensory nerve action potential amplitudes (axonal pattern), though demyelinating features with reduced conduction velocities have also been reported. Electromyography may reveal denervation changes in distal muscles if motor axon loss has occurred.
- Differential diagnosis: B12 deficiency neuropathy must be distinguished from other common causes of peripheral neuropathy including diabetic neuropathy, alcoholic neuropathy, chronic inflammatory demyelinating polyneuropathy (CIDP), and inherited neuropathies. B12 levels should be measured in any patient presenting with unexplained peripheral neuropathy.
Cognitive Impairment and Dementia
- Spectrum of cognitive dysfunction: B12 deficiency can produce a range of cognitive impairments from mild difficulty with concentration and memory to severe dementia with personality changes, psychosis, and delirium. Historically, B12 deficiency-associated dementia was termed “megaloblastic madness,” though cognitive impairment can occur without hematological abnormalities.
- Mechanisms of cognitive impairment: Cognitive decline in B12 deficiency results from white matter demyelination (disrupting cortical connectivity), hippocampal neurotoxicity from elevated homocysteine (impairing memory consolidation), impaired neurotransmitter synthesis (affecting mood and executive function), and cerebrovascular disease from homocysteine-mediated endothelial damage (contributing to vascular cognitive impairment).
- Relationship to Alzheimer’s disease: Low B12 status and elevated homocysteine have been associated with increased risk of Alzheimer’s disease in prospective cohort studies. Whether B12 deficiency directly contributes to Alzheimer’s pathology or represents a modifiable risk factor that lowers the clinical threshold for manifesting dementia in individuals with pre-existing amyloid and tau pathology is actively investigated.
- Reversibility: Cognitive impairment due to B12 deficiency is potentially reversible when treated early, though recovery is typically slow (months to years) and incomplete when deficiency has been prolonged. The degree of recovery is inversely related to the duration and severity of deficiency before treatment.
Optic Neuropathy
- Clinical features: B12 deficiency can cause bilateral optic neuropathy presenting as progressive, painless, symmetric visual loss with central or centrocecal scotomas. Visual acuity may range from mild reduction to severe impairment. The optic disc may appear normal (retrobulbar optic neuropathy) or show pallor reflecting optic atrophy in chronic cases.
- Mechanism: The optic nerve, as a central nervous system white matter tract, is vulnerable to the same demyelinating and metabolic processes that affect the spinal cord in SCD. The papillomacular bundle, responsible for central visual acuity and color vision, is preferentially affected, possibly due to its high metabolic demands.
- Differential diagnosis: B12 deficiency optic neuropathy must be distinguished from other nutritional optic neuropathies (folate, copper, thiamine), toxic optic neuropathy (ethambutol, methanol, tobacco-alcohol amblyopia), hereditary optic neuropathies (Leber’s), and compressive lesions. Bilateral, symmetric, painless visual loss with centrocecal scotomas should prompt B12 measurement.
Autonomic Neuropathy
- Manifestations: Though less commonly recognized than sensory or motor neuropathy, B12 deficiency can affect the autonomic nervous system. Reported autonomic manifestations include orthostatic hypotension, urinary incontinence or retention, erectile dysfunction, gastroparesis, and anhidrosis (reduced sweating) or hyperhidrosis.
- Bladder dysfunction: Urinary symptoms are particularly notable in B12 deficiency, occurring in up to 25% of patients with SCD. Neurogenic bladder from B12 deficiency can present as urinary urgency, frequency, incontinence, or retention, depending on the level and extent of spinal cord involvement.
- Cardiovascular autonomic dysfunction: Orthostatic hypotension and resting tachycardia have been reported in B12-deficient patients, attributed to impaired baroreceptor reflex function and sympathetic vasomotor dysfunction. Autonomic testing may reveal reduced heart rate variability and abnormal blood pressure responses to standing.
B12 and Multiple Sclerosis Research
- Overlapping clinical features: B12 deficiency and multiple sclerosis (MS) share several clinical and radiological features, including spinal cord demyelination, Lhermitte sign, optic neuropathy, and white matter lesions on MRI. This overlap necessitates ruling out B12 deficiency in any patient being evaluated for MS, as B12 deficiency is a treatable mimic.
- Prevalence of B12 deficiency in MS: Several studies have reported a higher prevalence of low B12 levels in MS patients compared to healthy controls, though whether this represents a causal relationship, a consequence of the disease, or a coincidental finding remains unclear.
- Remyelination potential: Given B12’s critical role in myelin synthesis, researchers have investigated whether B12 supplementation might promote remyelination in MS. While ensuring B12 adequacy is important for MS patients, clinical trials have not demonstrated that B12 supplementation above normal levels provides additional therapeutic benefit in MS.
- High-dose biotin in MS: Interestingly, high-dose biotin (vitamin B7, 300 mg daily) has been investigated as a treatment for progressive MS, with the rationale that it may support myelin synthesis through enhanced fatty acid production. While initial trials showed some promise, larger confirmatory trials have yielded disappointing results.
Methylcobalamin vs. Cyanocobalamin for Neurological Conditions
- Cyanocobalamin: This is the most common synthetic form of B12, widely used in supplements and fortified foods. It requires conversion to its active coenzyme forms (methylcobalamin and adenosylcobalamin) in the body. Cyanocobalamin is inexpensive, highly stable, and has the most extensive safety and efficacy data.
- Methylcobalamin: This is the active coenzyme form for the methionine synthase reaction. Proponents argue that methylcobalamin may be superior for neurological conditions because it directly provides the active form needed for CNS methylation reactions, potentially bypasses metabolic conversion steps that may be impaired, and may achieve higher concentrations in the CNS and peripheral nerves.
- Clinical evidence: Japanese clinical studies (where methylcobalamin has been used therapeutically for decades) have reported benefits of high-dose methylcobalamin (1,500 mcg daily) in peripheral neuropathy, with improvements in nerve conduction velocities and sensory symptoms. However, head-to-head comparison trials against cyanocobalamin are limited and generally of modest quality.
- Adenosylcobalamin: This active coenzyme form is required for the methylmalonyl-CoA mutase reaction and is more directly relevant to the prevention of abnormal fatty acid incorporation into myelin. Combination supplements providing both methylcobalamin and adenosylcobalamin are available but lack robust clinical trial support.
- Practical recommendation: For most patients with B12 deficiency neuropathy or myelopathy, the most important factor is prompt and adequate B12 replacement rather than the specific form used. Cyanocobalamin remains the standard of care in most guidelines. Methylcobalamin may be a reasonable alternative, particularly for patients with documented neurological involvement, but should not delay initiation of standard treatment.
Clinical Recognition and Treatment
- Early recognition: The key to preventing permanent neurological damage from B12 deficiency is early recognition. Any patient presenting with unexplained neuropathy, myelopathy, cognitive decline, or psychiatric symptoms should have B12 levels measured. The classic triad of anemia, glossitis, and neuropathy is present in fewer than 30% of B12-deficient patients; neurological symptoms can precede hematological abnormalities by months to years.
- Emergency treatment: Patients with acute neurological deterioration from B12 deficiency (progressive myelopathy, rapidly worsening neuropathy) should receive urgent parenteral B12 replacement. A typical emergency protocol involves intramuscular cyanocobalamin or hydroxocobalamin 1,000 mcg daily for 5 to 7 days, followed by weekly injections for 4 weeks, then monthly injections indefinitely.
- Avoiding folate masking: Folic acid supplementation without concurrent B12 can partially correct the megaloblastic anemia of B12 deficiency while allowing neurological damage to progress unchecked. This “masking” effect is a critical safety concern, particularly in countries with mandatory folic acid fortification. B12 status should be assessed before initiating folate therapy in patients at risk for B12 deficiency.
- Monitoring neurological recovery: Neurological recovery after B12 replacement should be monitored with serial neurological examinations, including assessment of vibration sense, proprioception, reflexes, gait, and cognitive function. Nerve conduction studies can be repeated at 6 to 12 months to document electrophysiological improvement. MRI of the spinal cord should be repeated if initial imaging was abnormal.
- Prognosis: With prompt treatment, most patients experience significant neurological improvement. Sensory symptoms typically improve before motor deficits. Complete recovery is most likely when treatment is initiated within 3 months of symptom onset. Patients with symptoms persisting for more than 12 months before treatment are likely to have residual neurological deficits.
Summary
Vitamin B12 is indispensable for nervous system health through its roles in myelin synthesis, SAMe-dependent methylation, and normal neuronal metabolism. Deficiency produces a wide range of neurological manifestations including peripheral neuropathy, subacute combined degeneration of the spinal cord, cognitive impairment, optic neuropathy, and autonomic dysfunction. Early recognition and prompt treatment are essential, as neurological damage becomes increasingly irreversible with prolonged deficiency. B12 levels should be measured in any patient with unexplained neurological symptoms, and treatment should not be delayed pending confirmatory testing when clinical suspicion is high.
This content is provided for informational purposes only and does not constitute medical advice. Consult a qualified healthcare provider for the diagnosis and treatment of any suspected vitamin deficiency, particularly when neurological symptoms are present.
Research Papers and References
The following are landmark and frequently cited research papers underpinning the claims on this page. Links resolve to the publisher DOI or PubMed record.
Foundational Clinical Reviews
- Stabler SP. Clinical practice. Vitamin B12 deficiency. New England Journal of Medicine. 2013;368(2):149-160.
- Green R, Allen LH, Bjorke-Monsen AL, et al. Vitamin B12 deficiency. Nature Reviews Disease Primers. 2017;3:17040.
- Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226.
B12 and the Nervous System
- Reynolds E. Vitamin B12, folic acid, and the nervous system. Lancet Neurology. 2006;5(11):949-960.
- PubMed — Subacute combined degeneration and B12 MRI findings (topic search)
- PubMed — B12 peripheral neuropathy and nerve conduction studies (topic search)
Cognition, Homocysteine, and Dementia
- PubMed — B12, homocysteine, and cognitive decline (topic search)
- PubMed — B12 and Alzheimer’s disease prospective cohorts (topic search)
Methylcobalamin and Neuropathy Trials
- PubMed — Methylcobalamin and peripheral neuropathy RCTs (topic search)
- PubMed — Cyanocobalamin vs. methylcobalamin head-to-head (topic search)
Optic Neuropathy and Autonomic Dysfunction
- PubMed — B12 deficiency and bilateral optic neuropathy (topic search)
- PubMed — B12 deficiency and autonomic dysfunction (topic search)
External Authoritative Resources
- NIH Office of Dietary Supplements — Vitamin B12 Fact Sheet for Health Professionals
- Linus Pauling Institute — Micronutrient Information Center: Vitamin B12
- Harvard T.H. Chan School of Public Health — The Nutrition Source: Vitamin B12
- PubMed — B12 deficiency and neurological manifestations (topic search)
Connections
- All Vitamins
- Vitamin B12’s Impossible Journey — interactive animation
- Vitamin B12
- B12 Deficiency Diagnosis
- Vitamin B9
- Vitamin B6
- Vitamin D3
- Vitamin E
- Methionine
- Anemia
- Alzheimer's Disease
- Peripheral Neuropathy
- Dementia
- Depression
- Iron
- Zinc
- Homocysteine
- Vitamin B12 Test
- Multiple Sclerosis