Molybdenum for Detoxification
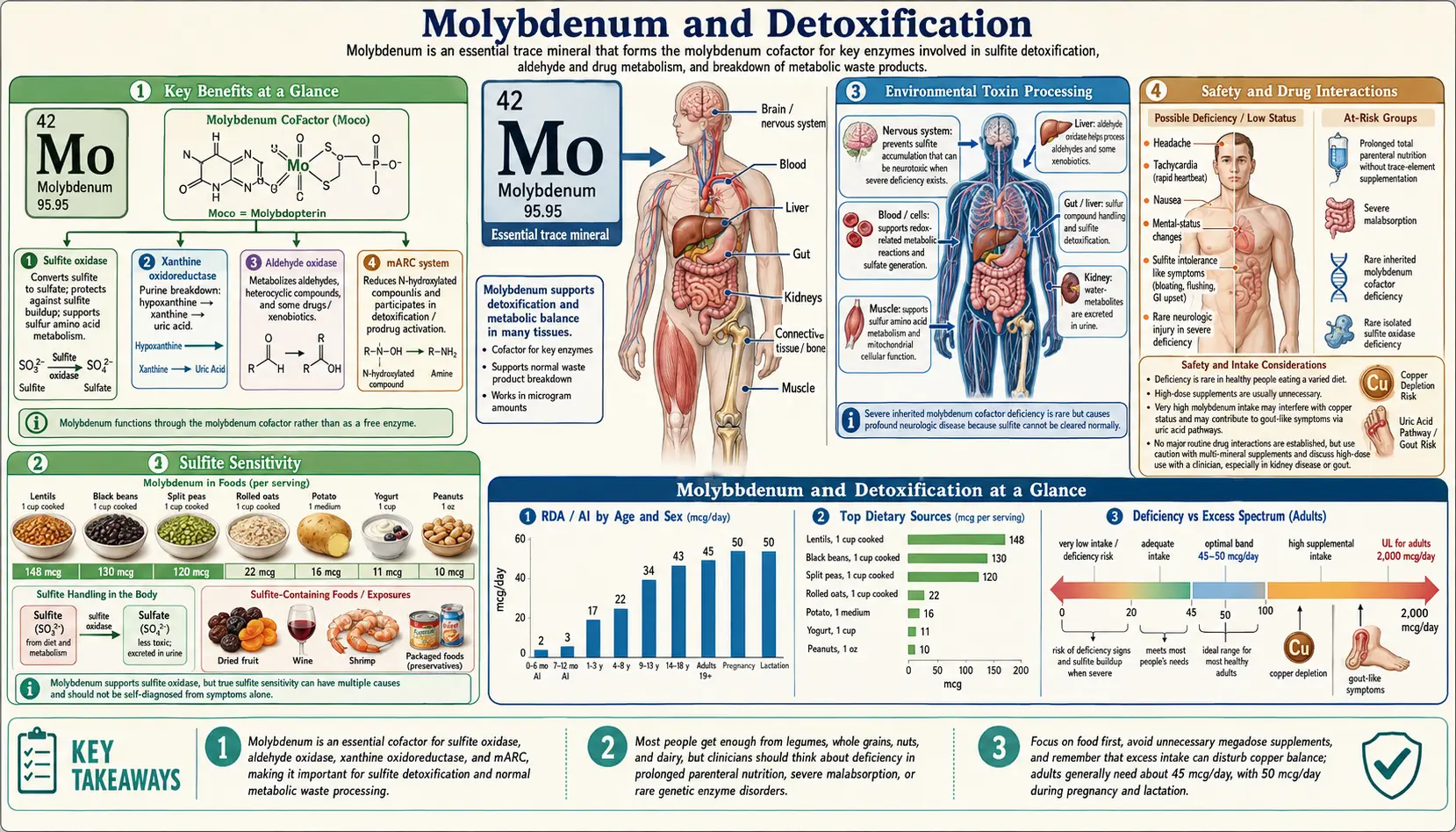
Molybdenum is the active center of three of the body's most consequential detoxification enzymes: sulfite oxidase (the only enzymatic pathway for converting toxic sulfite to inert sulfate), aldehyde oxidase (a broad-spectrum oxidizer of environmental aldehydes and pharmaceutical xenobiotics that operates independently of cytochrome P450), and xanthine oxidase (the terminal enzyme of purine catabolism, producing uric acid). Without functional molybdopterin cofactor, all three enzymes are inactive — a state seen in the catastrophic neonatal disease molybdenum cofactor deficiency, where sulfite accumulation alone is sufficient to cause overwhelming brain destruction within days of birth. This deep-dive maps each of the three molybdoenzymes, the toxic substrates they handle, the clinical disorders that arise when they fail, and the practical implications for adults with sulfite sensitivity, gout, drug-metabolism considerations, or significant xenobiotic exposure.
Table of Contents
- The Three Molybdoenzymes of Detoxification
- Sulfite Oxidase and Sulfite Detoxification
- Sulfite Sensitivity (Asthma, Wine Reactions)
- Aldehyde Oxidase and Xenobiotic Metabolism
- Drug Metabolism (Pharmaceutical Substrates)
- Xanthine Oxidase and Purine Catabolism
- Environmental Toxin Processing
- Molybdenum Cofactor Deficiency and Fosdenopterin
- Clinical Relevance for Adults
- Dosing and Dietary Sources
- Cautions and Drug Interactions
- Key Research Papers
- Connections
- Featured Videos
The Three Molybdoenzymes of Detoxification
Despite the body's tiny absolute requirement for molybdenum (45 mcg/day adult RDA, less than one ten-thousandth of the daily protein requirement), this trace mineral is irreplaceable because it is the active center of three distinct enzymes that collectively handle a remarkably broad portfolio of toxic substrates. Each enzyme uses the same molybdopterin cofactor (Moco), but each is tuned for a different substrate class:
- Sulfite oxidase — mitochondrial intermembrane space enzyme that converts sulfite (SO32−) to sulfate (SO42−). This is the sole enzymatic pathway for sulfite elimination; there is no backup. See the Sulfite Metabolism deep-dive.
- Aldehyde oxidase (AOX1) — cytosolic molybdoflavoprotein expressed predominantly in liver, also lung, kidney, gut, and brain. Oxidizes aldehydes, nitrogen-containing heterocycles, and many pharmaceuticals that bypass the cytochrome P450 system.
- Xanthine oxidase (XO) — catalyzes hypoxanthine → xanthine → uric acid, the terminal two steps of purine catabolism. Generates superoxide and hydrogen peroxide as byproducts. See the Uric Acid deep-dive.
A fourth molybdoenzyme, mitochondrial amidoxime reducing component (mARC), was characterized more recently and reduces N-hydroxylated substrates (including drug prodrugs and possibly endogenous N-hydroxyl metabolites). Its full physiological role is still being mapped.
Critically, all four enzymes share the same biosynthetic machinery for the molybdopterin cofactor — if cofactor biosynthesis fails, every molybdoenzyme fails simultaneously. This is why molybdenum cofactor deficiency is so catastrophic.
Sulfite Oxidase and Sulfite Detoxification
Sulfite oxidase resides in the mitochondrial intermembrane space and catalyzes the oxidation of sulfite (SO32−) to sulfate (SO42−), using cytochrome c as the physiological electron acceptor. This reaction represents the sole enzymatic pathway for sulfite elimination in humans. There is no redundant or compensatory mechanism — if sulfite oxidase fails, sulfite accumulates to toxic levels.
Endogenous sulfite is continuously generated from the catabolism of the sulfur amino acids methionine and cysteine. Degradation of cysteine through the cysteine sulfinic acid pathway yields sulfite as a penultimate intermediate before conversion to sulfate. A typical adult metabolizes roughly 2.5–3 grams of sulfur amino acids daily, generating substantial sulfite that must be cleared.
Exogenous sulfites are ingested through foods and beverages preserved with sulfiting agents (sodium sulfite, sodium bisulfite, sodium metabisulfite, potassium bisulfite, potassium metabisulfite, and sulfur dioxide). Common sources include wine, beer, dried fruits, fruit juices, processed meats, and condiments. The total daily intake from a typical Western diet ranges from 10–180 mg of sulfite equivalents.
Sulfite toxicity at the cellular level involves multiple mechanisms: disruption of disulfide bonds in proteins leading to loss of tertiary structure; reaction with S-adenosylmethionine and other sulfonium compounds; generation of sulfite radical anions (SO3−) through one-electron oxidation that damage lipid membranes, DNA, and mitochondrial components; and depletion of glutathione stores via reaction with cysteinyl residues.
The nervous system is exquisitely sensitive to sulfite accumulation. Sulfite interferes with NMDA receptor function, disrupts calcium homeostasis, causes mitochondrial energy failure in neurons, and induces apoptotic cell death in developing brain tissue. This neuronal vulnerability is the dominant clinical feature of sulfite oxidase deficiency.
For the full molecular and clinical picture — including isolated sulfite oxidase deficiency, the role of dietary sulfite restriction, and how molybdenum supplementation may support enzymatic capacity — see the dedicated Sulfite Metabolism page.
Sulfite Sensitivity (Asthma, Wine Reactions)
Sulfite sensitivity affects an estimated 1% of the general population and up to 5–10% of individuals with asthma, particularly those with steroid-dependent disease. The phenotype is dose-dependent and varies dramatically between individuals: some people tolerate sulfite-rich foods without symptoms, while others react to a single glass of preserved wine.
Symptoms of sulfite sensitivity include:
- Bronchospasm — the classic respiratory presentation, sometimes severe enough to require emergency treatment
- Wheezing, cough, and chest tightness within minutes of ingestion
- Urticaria and angioedema (especially around the lips and face)
- Gastrointestinal cramping and diarrhea
- Hypotension, flushing, and tachycardia
- In severe cases, anaphylactoid reactions requiring epinephrine
The mechanism is thought to involve insufficient sulfite oxidase capacity relative to the sulfite load, resulting in transient sulfite accumulation that triggers mast cell degranulation, parasympathetic nerve stimulation in airways, and direct airway smooth muscle contraction. The variability between individuals likely reflects polymorphisms in sulfite oxidase (SUOX gene) and in cofactor biosynthesis enzymes, plus differences in dietary methionine load and gut sulfate-reducing bacteria.
Individuals with sulfite sensitivity may benefit from ensuring adequate molybdenum intake to optimize sulfite oxidase function, though randomized clinical trial data on molybdenum supplementation for sulfite sensitivity remain limited. Practical strategies include reading labels (sulfites must be declared above 10 ppm), avoiding the highest-sulfite foods (preserved wine, dried apricots, packaged shrimp), and discussing rescue inhaler use with the prescribing pulmonologist.
Regulatory agencies require sulfite labeling on foods containing more than 10 parts per million of sulfite equivalents due to the clinical significance of sulfite-induced reactions — this 1986 FDA rule followed multiple deaths from sulfite-triggered asthma attacks in salad bar settings.
Aldehyde Oxidase and Xenobiotic Metabolism
Aldehyde oxidase (AOX1) is a cytosolic molybdoflavoprotein expressed predominantly in the liver, with additional expression in the lungs, kidneys, gastrointestinal tract, and brain. AOX1 contains a molybdopterin cofactor, a FAD cofactor, and two iron-sulfur (2Fe-2S) clusters, enabling it to catalyze the oxidation of a remarkably broad range of substrates.
Endogenous substrates include retinaldehyde (converted to retinoic acid), pyridoxal (vitamin B6 aldehyde form), and various biogenic aldehydes produced during neurotransmitter metabolism — including the aldehyde intermediates of dopamine and serotonin degradation. AOX1 thus contributes to neurotransmitter turnover in addition to its xenobiotic role.
Xenobiotic metabolism: AOX1 oxidizes a wide spectrum of exogenous compounds including:
- Nitrogen-containing heterocycles (pyridines, pyrimidines, purines, quinolines, phthalazines)
- Aromatic and aliphatic aldehydes
- Nitro-compounds
- Environmental aldehydes from tobacco smoke, vehicle exhaust, and industrial emissions
- Lipid peroxidation products including 4-hydroxynonenal and malondialdehyde
This broad substrate range makes AOX1 a significant enzyme in phase I drug metabolism, complementing the cytochrome P450 system. Unlike P450, AOX1 does not require NADPH or molecular oxygen as a co-substrate — it uses water as the oxygen donor and produces hydrogen peroxide as a byproduct.
AOX1 also contributes to the detoxification of environmental aldehydes encountered through tobacco smoke, air pollution, and industrial chemical exposure. Populations with high inhalation aldehyde exposure may place increased demand on this pathway.
Drug Metabolism (Pharmaceutical Substrates)
AOX1-mediated metabolism has become a major consideration in drug development, as compounds cleared primarily by aldehyde oxidase may exhibit species-dependent pharmacokinetics (AOX1 activity varies significantly between humans, rodents, and other preclinical species), leading to unexpected drug failures in clinical trials.
Pharmaceutical substrates of aldehyde oxidase include:
- Famciclovir — antiviral prodrug, activated by AOX1 to penciclovir for treatment of herpes simplex and zoster
- Zaleplon — sedative-hypnotic, AOX1 is the primary metabolic pathway
- Methotrexate — converted to 7-hydroxymethotrexate, which retains some activity and contributes to nephrotoxicity at high doses
- 6-Mercaptopurine and azathioprine — partial AOX1 contribution (xanthine oxidase is the dominant pathway)
- Ziprasidone — atypical antipsychotic, partially metabolized by AOX1
- Idelalisib and several kinase inhibitors used in oncology
- SGX523, BIBX1382, and other investigational kinase inhibitors that were withdrawn from development due to unexpectedly rapid AOX1-mediated clearance discovered only in human pharmacokinetic studies
The clinical implication is that genetic polymorphisms in AOX1 can affect drug metabolism rates, with some variants producing rapid or slow metabolizer phenotypes that influence the efficacy and toxicity of AO-substrate drugs. Patients taking multiple AOX1-substrate medications, or with documented AOX1 variants, may require dose adjustment.
Xanthine Oxidase and Purine Catabolism
Xanthine oxidase (XO) and its NAD+-dependent form xanthine dehydrogenase (XDH) catalyze the final two steps of purine catabolism: hypoxanthine to xanthine, and xanthine to uric acid. The same protein cycles between the two forms (XDH is the dominant form under physiological conditions; XO is generated by proteolytic or oxidative conversion during inflammation, ischemia, or tissue injury).
This represents a detoxification function because hypoxanthine and xanthine are relatively insoluble and must be converted to uric acid for renal excretion. In their accumulated form (in enzyme deficiency), they can precipitate in renal tubules and cause obstructive nephropathy and xanthine kidney stones.
Xanthine oxidase uses molecular oxygen as an electron acceptor, producing superoxide anion (O2−) and hydrogen peroxide (H2O2) as byproducts. While these reactive oxygen species serve antimicrobial functions, their overproduction contributes to tissue damage, particularly in ischemia-reperfusion injury.
In the context of detoxification, XO contributes to the oxidative metabolism of various purines and pteridines beyond the canonical hypoxanthine-xanthine pathway, including the metabolism of certain purine analog drugs (6-mercaptopurine, azathioprine).
XO is released into the circulation during tissue injury and inflammation (particularly from the liver and intestinal mucosa), where it binds to vascular endothelium via glycosaminoglycans and can generate oxidative stress at the vessel wall, contributing to ischemia-reperfusion injury and endothelial dysfunction.
Xanthine oxidase inhibitors (allopurinol, febuxostat) are used therapeutically to reduce uric acid production in gout and to limit oxidative damage in conditions such as tumor lysis syndrome and ischemia-reperfusion injury. For a deeper exploration of XO biology, uric acid as antioxidant, gout pharmacology, and the high-purine diet interaction, see the dedicated Uric Acid page.
Environmental Toxin Processing
Molybdenum-dependent enzymes collectively provide a broad-spectrum detoxification capacity that complements the cytochrome P450 and conjugation enzyme systems. Several environmental exposures place specific demands on this pathway:
- Sulfur dioxide (SO2) exposure from air pollution and fossil fuel combustion generates sulfite in the respiratory tract, which must be detoxified by sulfite oxidase. Populations in heavy industrial areas, near coal-fired power plants, or with significant tobacco smoke exposure may place increased demand on this pathway.
- Aldehyde pollutants such as formaldehyde (from particle board, foam insulation, and tobacco smoke), acetaldehyde (from alcohol metabolism and air pollution), and acrolein (from cooking oils heated to smoke point, vehicle exhaust, and industrial emissions) are substrates for aldehyde oxidase-mediated oxidation.
- The purine load from high dietary intake of nucleic acid-rich foods (organ meats, sardines, anchovies, yeast extracts, beer) requires adequate xanthine oxidase activity for efficient catabolism and excretion.
- Mold exposure — some integrative practitioners suggest molybdenum supplementation supports detoxification of acetaldehyde produced by Candida overgrowth and mycotoxin exposure, though high-quality clinical evidence is limited.
- Tobacco smoke contains both inhaled sulfur dioxide and a complex mixture of aldehyde compounds — both substrate classes for molybdenum-dependent enzymes.
Molybdenum status may therefore be particularly relevant for individuals with high xenobiotic exposure, heavy dietary sulfite intake, significant purine loads, or occupational exposure to aldehydes and sulfur compounds. The 45 mcg/day RDA reflects minimum requirements for asymptomatic adults; functional optimization for high-exposure populations may benefit from higher intake within the safe range.
Molybdenum Cofactor Deficiency and Fosdenopterin
Molybdenum cofactor deficiency (MoCD) is the most dramatic demonstration of molybdenum's detoxification importance. This rare autosomal recessive disorder (estimated incidence 1 in 100,000 to 1 in 200,000 live births) results from mutations in the cofactor biosynthesis genes MOCS1, MOCS2, MOCS3, or GPHN. Loss of cofactor production simultaneously inactivates all three molybdoenzymes — sulfite oxidase, xanthine oxidase, and aldehyde oxidase — though sulfite toxicity is the primary driver of clinical devastation.
The clinical presentation:
- Intractable neonatal seizures within hours to days of birth
- Severe encephalopathy with apnea and feeding difficulty
- Lens subluxation visible on slit-lamp examination (the lens dislocates because sulfite damages the zonular fibers anchoring it)
- Progressive cystic destruction of cerebral white matter visible on MRI within weeks
- Microcephaly developing over the first months
- Death typically within the first year if untreated
Biochemically, patients exhibit markedly elevated urinary sulfite (detectable by reagent strip), S-sulfocysteine, thiosulfate, and decreased sulfate excretion, plus elevated xanthine and decreased uric acid from the concurrent xanthine oxidase failure.
Cyclic pyranopterin monophosphate (cPMP, fosdenopterin, brand name Nulibry) replacement therapy for MoCD type A (MOCS1 mutations) represents a breakthrough treatment. cPMP is the precursor that the MOCS1 gene product would normally make; supplying it exogenously rescues cofactor biosynthesis and restores all three molybdoenzyme activities. Daily intravenous infusion, initiated within days of birth, dramatically reduces seizure burden, prevents progressive neurological damage, and produces near-normal developmental outcomes in early-treated infants. FDA approval came in February 2021. Treatment is lifelong and costs approximately $1 million per year per patient.
MoCD type B (MOCS2) and type C (MOCS3) currently lack approved replacement therapy because the relevant biosynthetic intermediates are less stable; experimental approaches are under investigation.
Isolated sulfite oxidase deficiency (SUOX gene mutations) is clinically indistinguishable from MoCD but affects only sulfite oxidase — xanthine oxidase and aldehyde oxidase function normally. The clinical picture is dominated by sulfite-driven neurological devastation. No specific replacement therapy exists; aggressive dietary sulfur restriction provides only partial benefit.
Clinical Relevance for Adults
Although frank molybdenum deficiency is exceedingly rare in individuals consuming oral diets, suboptimal molybdenum status in the context of high sulfite exposure, heavy purine intake, or significant xenobiotic burden may have clinically relevant effects on detoxification capacity. Adult populations where molybdenum status warrants attention:
- Long-term total parenteral nutrition (TPN) — the only consistently documented setting for adult molybdenum deficiency. A single landmark case (Abumrad et al. 1981) presented with tachycardia, tachypnea, mental status changes, scotomata, low serum methionine and uric acid, and high urinary sulfite, with resolution after molybdenum addition to the TPN formula. All TPN formulations now include molybdenum (typically as ammonium molybdate).
- Sulfite-sensitive asthma — ensuring adequate molybdenum intake supports sulfite oxidase capacity; clinical benefit is plausible though not proven by RCTs.
- Hepatic disease — reduced enzyme expression and cofactor availability can impair molybdenum enzyme function, potentially compounding the detoxification deficit already present from impaired cytochrome P450 activity in liver disease.
- Significant tobacco smoke or air pollution exposure — high inhaled sulfur dioxide and aldehyde loads place increased demand on the molybdenum-dependent detoxification pathway.
- Mold and mycotoxin exposure — integrative protocols frequently include molybdenum to support acetaldehyde clearance, though high-quality clinical evidence is limited.
- Patients on multiple AOX1-substrate medications — ensuring cofactor availability supports drug-metabolism capacity.
For adults in these contexts, intake of 75–500 mcg/day from food plus low-dose supplement is generally appropriate and well within the 2,000 mcg/day tolerable upper intake limit.
Dosing and Dietary Sources
- RDA (adults) — 45 mcg/day; pregnancy and lactation 50 mcg/day
- Tolerable Upper Intake Level (UL) — 2,000 mcg/day (2 mg/day) for adults
- Legumes — black-eyed peas, lima beans, kidney beans, lentils are the richest sources (a single cup of cooked black-eyed peas provides ~200 mcg, well over the RDA)
- Whole grains — oats, barley, whole wheat (~25–50 mcg per cup)
- Nuts and seeds — almonds, peanuts, sunflower seeds
- Organ meats — liver and kidney (highly variable depending on soil)
- Typical supplements — 50–500 mcg/day as sodium molybdate or ammonium molybdate; molybdenum glycinate chelate is also available
- Soil molybdenum content varies dramatically — geographic regions with molybdenum-poor soils (parts of China's Linxian County, parts of South Africa) historically had elevated rates of esophageal cancer and other conditions plausibly linked to low molybdenum status, though confounding factors (selenium, zinc, nitrate) complicate the picture
Cautions and Drug Interactions
- Copper antagonism — High molybdenum intake (especially combined with sulfur) can induce functional copper deficiency through formation of thiomolybdate complexes that bind copper irreversibly. This relationship is exploited therapeutically: tetrathiomolybdate is used to decopper patients with Wilson disease. The clinical implication is that supplementation above 500 mcg/day should be balanced with adequate copper intake (and monitored if continued for months).
- Gout — Very high intakes (>500 mcg/day chronically) have been associated with elevated uric acid in occupational exposure reports from molybdenum mining and processing regions. Patients with established gout should keep intake near the RDA unless directed otherwise.
- Allopurinol / febuxostat — These xanthine oxidase inhibitors interact pharmacodynamically with the enzyme; clinical impact of dietary molybdenum is minimal but theoretically supplementation could marginally accelerate uric acid generation.
- 6-Mercaptopurine / azathioprine — XO metabolism relevant; discuss with prescribing physician before adding molybdenum supplements.
- TPN patients — require formal molybdenum supplementation in total parenteral nutrition per ASPEN guidelines.
- Pregnancy — RDA increases modestly to 50 mcg/day; high-dose supplementation is not recommended without specific clinical indication.
This content is provided for informational purposes only and does not constitute medical advice. Consult a qualified healthcare provider before starting molybdenum supplementation.
Key Research Papers
- Hille R, Hall J, Basu P (2014). The mononuclear molybdenum enzymes. Chemical Reviews. — PubMed
- Schwarz G, Mendel RR, Ribbe MW (2009). Molybdenum cofactors, enzymes and pathways. Nature. — PubMed
- Schwahn BC et al. (2015). Efficacy and safety of cyclic pyranopterin monophosphate in severe molybdenum cofactor deficiency type A. The Lancet. — PubMed
- Reiss J, Hahnewald R (2011). Molybdenum cofactor deficiency: mutations in GPHN, MOCS1, and MOCS2. Human Mutation. — PubMed
- Pryde DC et al. (2010). Aldehyde oxidase: an enzyme of emerging importance in drug discovery. Journal of Medicinal Chemistry. — PubMed
- Harrison R (2004). Physiological roles of xanthine oxidoreductase. Drug Metabolism Reviews. — PubMed
- Kisker C, Schindelin H, Rees DC (1997). Molybdenum-cofactor-containing enzymes: structure and mechanism. Annual Review of Biochemistry. — PubMed
- Turnlund JR (2002). Molybdenum metabolism and requirements in humans. Metal Ions in Biological Systems. — PubMed
- Abumrad NN et al. (1981). Amino acid intolerance during prolonged total parenteral nutrition reversed by molybdate therapy. American Journal of Clinical Nutrition. — PubMed
- Mendel RR (2013). The molybdenum cofactor. Journal of Biological Chemistry. — PubMed
- Bender D et al. (2019). Impaired mitochondrial maturation of sulfite oxidase in a patient with severe isolated SUOX deficiency. Human Molecular Genetics. — PubMed
- Garattini E, Terao M (2012). The role of aldehyde oxidase in drug metabolism. Expert Opinion on Drug Metabolism & Toxicology. — PubMed
PubMed Topic Searches
- PubMed: Sulfite oxidase deficiency
- PubMed: Molybdenum cofactor deficiency
- PubMed: Aldehyde oxidase drug metabolism
- PubMed: Xanthine oxidase, uric acid, gout
- PubMed: Molybdopterin biosynthesis
Connections
- Molybdenum Benefits Hub
- Molybdenum Overview
- Molybdenum for Sulfite Metabolism
- Molybdenum for Uric Acid
- Molybdenum for Iron Utilization
- Sulfur
- Cysteine
- Methionine
- Copper
- Gout
- Sulfites
- Selenium
- Zinc
- Liver Cleansing
- Gerson Detoxification
- Uric Acid Test
- Glutathione
- Oxidative Stress
- Organ Meats
- Asthma
- Liver Disease
- All Minerals