Molybdenum for Uric Acid
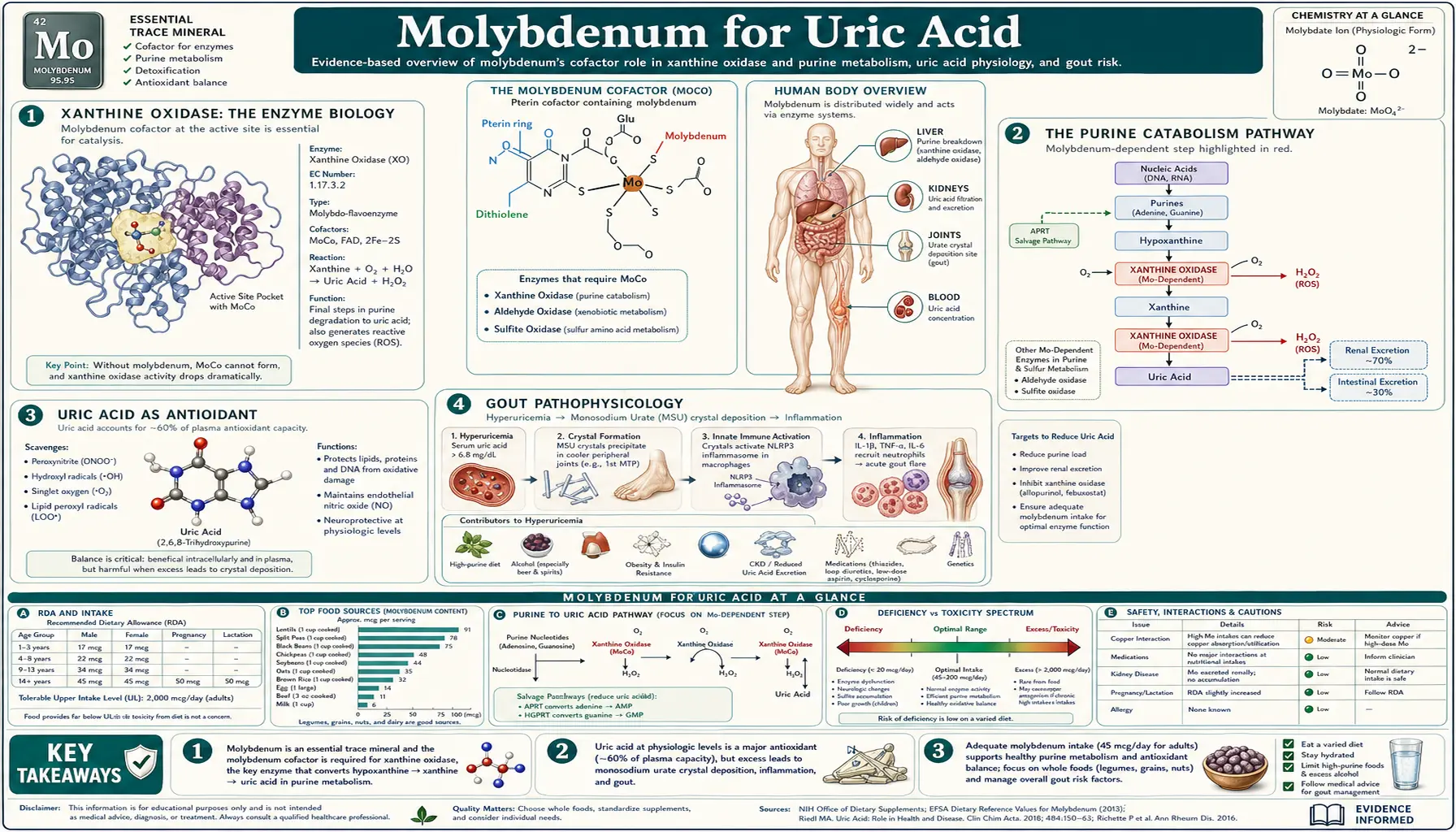
Xanthine oxidase, the second of the body's three molybdenum-dependent enzymes, catalyzes the terminal two steps of purine catabolism: hypoxanthine to xanthine, and xanthine to uric acid. This is the enzymatic target of allopurinol and febuxostat, the two pillars of gout pharmacotherapy. The biology is far more interesting than the simple "block uric acid production" framing: xanthine oxidase generates reactive oxygen species as catalytic byproducts (a major source of cardiovascular oxidative stress), uric acid itself is a powerful antioxidant providing roughly half the antioxidant capacity of human plasma, and the high-purine dietary inputs (organ meats, sardines, anchovies, beer, yeast extracts) interact with the molybdenum-dependent system in ways that matter for both gout prevention and general metabolic health. This deep-dive maps xanthine oxidase biology, purine sources, the gout connection, the underappreciated antioxidant role of uric acid, and the dietary and pharmacologic levers available for managing the system.
Table of Contents
- Xanthine Oxidase: The Enzyme Biology
- The Purine Catabolism Pathway
- Uric Acid as Antioxidant
- Gout Pathophysiology
- Allopurinol — The First-Line XO Inhibitor
- Febuxostat — The Second-Line XO Inhibitor
- High-Purine Diets and the Gout Triggers
- Xanthine Oxidase as Source of Reactive Oxygen Species
- Xanthinuria (Xanthine Oxidase Deficiency)
- Molybdenum, Copper, and Gout Risk
- Tumor Lysis Syndrome and Ischemia-Reperfusion
- Key Research Papers
- Connections
- Featured Videos
Xanthine Oxidase: The Enzyme Biology
Xanthine oxidoreductase (XOR) exists in two interconvertible forms that share the same protein backbone:
- Xanthine dehydrogenase (XDH) — the dominant form under physiological conditions. Uses NAD+ as the electron acceptor, producing NADH. Generates relatively little reactive oxygen species.
- Xanthine oxidase (XO) — generated from XDH by proteolytic cleavage (during inflammation, tissue injury, or ischemia) or by reversible oxidation of critical cysteine residues. Uses molecular oxygen as the electron acceptor, producing superoxide (O2−) and hydrogen peroxide (H2O2) as byproducts.
The same protein, the same catalytic mechanism on the substrate side, but very different output on the electron-acceptor side. The XDH-to-XO conversion during inflammation is a critical mechanism by which the body shifts purine catabolism from an essentially metabolic function to a source of reactive oxygen species useful for antimicrobial defense (and damaging when sustained or amplified).
Each XOR monomer contains four redox-active centers:
- A molybdopterin (Moco) center at the substrate-binding site, where hypoxanthine and xanthine are oxidized
- Two iron-sulfur clusters (2Fe-2S each) that shuttle electrons from the molybdenum center to the FAD
- A FAD (flavin adenine dinucleotide) center where electrons are passed to the final electron acceptor (NAD+ in XDH form, O2 in XO form)
XOR is expressed primarily in liver and small intestinal epithelium, with lower levels in mammary gland, heart, brain, and vascular endothelium. The enzyme is released into circulation during tissue injury, where it binds to glycosaminoglycans on vascular endothelium and produces ROS at the vessel wall — a key mechanism in ischemia-reperfusion injury.
The Purine Catabolism Pathway
Purines (adenine, guanine, and their nucleotide and nucleoside derivatives) enter the catabolic pathway from three sources:
- Cellular nucleic acid turnover — the breakdown of DNA and RNA from dying cells releases purine nucleotides that must be either salvaged for reuse or catabolized
- Dietary purine intake — nucleic-acid-rich foods (organ meats, certain seafood, beer, yeast extracts) supply exogenous purines
- De novo purine synthesis — new purine production from amino acids, especially when cellular demand outstrips salvage
The catabolic pathway converges on a common intermediate, hypoxanthine, regardless of the source:
Adenosine → Inosine → Hypoxanthine [via adenosine deaminase, purine nucleoside phosphorylase]
Guanosine → Guanine → Xanthine [via purine nucleoside phosphorylase, guanase]
Hypoxanthine + O2 + H2O → Xanthine + H2O2 [xanthine oxidase, step 1]
Xanthine + O2 + H2O → Uric Acid + H2O2 [xanthine oxidase, step 2]
Uric acid is then excreted primarily through the kidneys (about two-thirds) and partly through the gut (about one-third). Renal handling involves filtration at the glomerulus followed by complex bidirectional transport in the proximal tubule (involving URAT1, GLUT9, ABCG2, and other transporters — many of which carry genetic variants that influence individual uric acid levels).
Humans and the great apes are unusual among mammals in lacking the enzyme uricase, which would otherwise convert uric acid further to the more soluble allantoin. The loss of uricase appears to have happened in primate evolution and is presumed to confer some adaptive benefit (most plausibly the antioxidant role discussed below) at the cost of vulnerability to hyperuricemia and gout when dietary purine intake is high.
Uric Acid as Antioxidant
Uric acid is not merely a metabolic waste product to be excreted. It serves as a major antioxidant in human plasma, accounting for approximately 50% of the total antioxidant capacity of blood when measured by standard assays. Uric acid scavenges:
- Peroxynitrite (ONOO−) — a particularly damaging species formed from nitric oxide and superoxide
- Hydroxyl radicals (·OH)
- Singlet oxygen
- Iron-driven peroxidation products
Uric acid is particularly important for protection against peroxynitrite, which is implicated in vascular endothelial damage, neurodegeneration, and inflammation. The evolutionary loss of uricase in primates plausibly conferred increased antioxidant capacity at the cost of gout susceptibility — a trade-off the primate lineage apparently found favorable.
This dual role of uric acid creates a clinical paradox:
- High uric acid (hyperuricemia) causes gout, contributes to kidney stones, and is associated with hypertension, metabolic syndrome, and cardiovascular disease
- Very low uric acid (hypouricemia, especially <2 mg/dL) is associated with increased risk of neurodegenerative disease (Parkinson's, multiple sclerosis, ALS), possibly due to loss of antioxidant protection
The optimal serum uric acid for general health appears to be in the middle range — somewhere around 3–5 mg/dL, well below the gout threshold (~6 mg/dL) but above the hypouricemia danger zone. Aggressive pharmacological uric acid lowering below 3 mg/dL with allopurinol or febuxostat may sacrifice some of the antioxidant benefit, though this is not currently a standard consideration in clinical gout management.
Gout Pathophysiology
Gout is the clinical syndrome of monosodium urate (MSU) crystal deposition in joints and soft tissues, with episodic acute inflammatory attacks and a long-term risk of chronic tophaceous arthritis.
The fundamental requirement is hyperuricemia — serum uric acid above the solubility limit (approximately 6.8 mg/dL at physiological pH and temperature). Above this concentration, urate crystallizes out as MSU monohydrate. Local factors (low temperature, low pH, mechanical stress) determine where crystals form: the first metatarsophalangeal joint (the classic gout target) is favored because it's the coolest large joint and bears mechanical load.
Hyperuricemia arises from two main causes:
- Overproduction (~10% of cases) — high purine intake, accelerated cellular turnover (psoriasis, hemolytic anemia, certain cancers, tumor lysis), genetic variants increasing purine synthesis
- Underexcretion (~90% of cases) — reduced renal clearance due to kidney disease, diuretic use, dehydration, or genetic variants in renal urate transporters (URAT1, GLUT9, ABCG2)
Acute gout attacks are triggered when MSU crystals are recognized by the NLRP3 inflammasome in tissue macrophages, releasing IL-1 beta and recruiting neutrophils to produce intense local inflammation. The classic features — sudden severe joint pain (often waking the patient from sleep), erythema, warmth, swelling — develop over hours to a single day. Acute attacks resolve over days with or without treatment.
Chronic tophaceous gout develops over years of sustained hyperuricemia, with MSU crystal masses (tophi) accumulating in soft tissue, particularly the ear helices, olecranon and prepatellar bursae, finger pads, and chronically inflamed joints. Tophi can erode bone and produce visible deformity.
Allopurinol — The First-Line XO Inhibitor
Allopurinol (first synthesized 1956, FDA-approved 1966) is a purine analog that binds the xanthine oxidase active site. It is metabolized by XO itself to oxypurinol, which then becomes the dominant inhibitor — oxypurinol binds the molybdenum center tightly and persistently, blocking further substrate access. This is a classic "suicide inhibitor" mechanism: the drug is activated by the enzyme it inhibits.
Allopurinol is the standard first-line urate-lowering therapy for chronic gout, with several characteristics:
- Dose — starting at 100 mg/day (or 50 mg in CKD), titrated up to a maximum of 800 mg/day to achieve target serum urate (typically <6 mg/dL, or <5 mg/dL in patients with tophi)
- Mechanism — reduces uric acid production by blocking conversion of hypoxanthine and xanthine to uric acid. Substrates accumulate proximally as hypoxanthine, which is more soluble than uric acid and can be salvaged back to nucleotides via HGPRT
- Onset — serum urate falls within days; clinical benefit on attack frequency takes 6–12 months as accumulated MSU crystal stores gradually dissolve
- Acute attack precipitation — paradoxically, allopurinol initiation can trigger acute gout attacks in the first 6 months by mobilizing crystals; prophylaxis with colchicine 0.5–1.2 mg/day or low-dose NSAID is standard
The major safety concern is allopurinol hypersensitivity syndrome (AHS), a rare but potentially fatal severe cutaneous adverse reaction (SCAR) including Stevens-Johnson syndrome and toxic epidermal necrolysis. Risk is strongly associated with the HLA-B*5801 allele, which is much more common in Han Chinese, Thai, and Korean populations than in European populations. HLA-B*5801 testing before allopurinol initiation is now recommended in high-prevalence populations.
Allopurinol also has clinically important interactions with azathioprine and 6-mercaptopurine: XO normally inactivates these drugs, and XO inhibition leads to dangerously high active drug levels with severe bone marrow suppression. The dose of azathioprine or 6-MP must be reduced by 50–75% (or the drug avoided entirely) when allopurinol is started.
Febuxostat — The Second-Line XO Inhibitor
Febuxostat (FDA-approved 2009) is a non-purine selective xanthine oxidase inhibitor. Unlike allopurinol, febuxostat is not a purine analog and inhibits XO by a different binding mode — it occupies the substrate channel and blocks substrate access without being itself metabolized.
Clinical role:
- Second-line urate-lowering therapy when allopurinol is contraindicated, not tolerated, or fails to achieve target urate at maximum tolerated dose
- Particularly useful in patients with HLA-B*5801 positivity (cross-reactive risk is much lower than with allopurinol)
- Dose: 40–80 mg/day; can be used in CKD without dose adjustment
- Effective even at modest doses, often achieving target urate in patients who failed standard allopurinol
A significant safety concern emerged from the CARES trial (Cardiovascular Safety of Febuxostat and Allopurinol in Patients With Gout and Cardiovascular Morbidities, White et al. 2018, NEJM): febuxostat was associated with significantly higher cardiovascular mortality compared to allopurinol in patients with established cardiovascular disease. The mechanism is uncertain. The FDA issued a boxed warning in 2019 limiting febuxostat to patients who have failed or cannot tolerate allopurinol.
High-Purine Diets and the Gout Triggers
Dietary purines contribute meaningfully to uric acid generation, though typically less than endogenous purine turnover. The highest-purine foods are:
- Organ meats — liver, kidney, sweetbreads (thymus, pancreas), brain. Organ meats can contain 500–1000 mg purines per 100 g serving, far above muscle meats.
- Small whole-eaten fish — sardines, anchovies, herring, smelt, mackerel. The DNA-rich tissue of the entire fish (not just muscle) drives high purine content. Salmon and trout are moderate.
- Game meats — venison and other wild game tend to be higher than farmed meats
- Yeast extracts — Marmite, Vegemite, brewer's yeast supplements. Concentrated yeast extracts can contain 1000+ mg purines per 100 g.
- Beer — not because of the alcohol but because of the yeast-derived purines. Beer raises uric acid more than equivalent alcohol amounts from wine or spirits.
- Mussels, scallops, crab, shrimp, lobster — moderate to high
- Mushrooms, asparagus, spinach, cauliflower — moderate plant purines, though epidemiology suggests plant purines have less gout-precipitating effect than animal purines
Modern epidemiology (Choi et al. NEJM 2004 and follow-up work) found that animal-source purines and fructose are the strongest dietary risk factors for incident gout, while plant-source purines, low-fat dairy, and vitamin C appear protective. The role of fructose is mechanistically interesting: fructose metabolism in the liver consumes ATP rapidly, leading to accumulation of AMP (adenosine monophosphate), which is then catabolized through the purine pathway to uric acid. High-fructose corn syrup and sugar-sweetened beverages are independent gout risk factors.
Alcohol raises uric acid through multiple mechanisms: direct stimulation of purine catabolism (similar to fructose), reduced renal urate excretion, and beer's yeast purine content. Wine in moderation appears to have less effect than beer or spirits.
For gout patients, dietary modification can reduce serum urate by approximately 1–1.5 mg/dL — meaningful but generally insufficient to reach target alone. Diet works best in combination with pharmacological urate-lowering therapy.
Xanthine Oxidase as Source of Reactive Oxygen Species
The XO form of the enzyme (generated from XDH during inflammation or tissue injury) produces superoxide and hydrogen peroxide as catalytic byproducts. This makes XO a quantitatively important source of vascular reactive oxygen species, with implications well beyond purine metabolism:
- Ischemia-reperfusion injury — the classic example. During ischemia (loss of blood flow), XDH is converted to XO and hypoxanthine accumulates because of ATP breakdown. When blood flow returns (reperfusion), the sudden oxygen supply allows XO to rapidly oxidize accumulated hypoxanthine, generating a burst of superoxide and H2O2 that damages reperfused tissue. This is a major mechanism in myocardial infarction, stroke, and organ transplantation injury.
- Endothelial dysfunction — XO bound to vascular endothelium contributes to oxidative stress at the vessel wall, impairing nitric oxide signaling and promoting atherosclerosis
- Heart failure — XO activity is elevated in heart failure and contributes to myocardial oxidative damage; the EXACT-HF and OPT-CHF trials of allopurinol/oxypurinol in heart failure showed mixed results but suggest potential benefit in selected patients
- Vascular calcification and hypertension — XO-derived ROS contribute to vascular smooth muscle proliferation and impaired vasodilation
The therapeutic implication is that allopurinol and febuxostat may have benefits beyond uric acid lowering — the ROS reduction from XO inhibition is potentially cardioprotective and neuroprotective. Several trials have explored allopurinol for cardiovascular outcomes, blood pressure, and neuroprotection in stroke, with promising but not conclusive results.
Xanthinuria (Xanthine Oxidase Deficiency)
Hereditary xanthinuria is a rare autosomal recessive disorder of xanthine oxidase function. Two types exist:
- Type I — isolated xanthine dehydrogenase deficiency (XDH gene mutations). Xanthine oxidase activity is absent; aldehyde oxidase and sulfite oxidase function normally.
- Type II — combined xanthine dehydrogenase and aldehyde oxidase deficiency (caused by mutations in the molybdenum cofactor sulfurase gene MOCOS, which is required for the terminal sulfuration step of both XDH and AOX1 cofactor; sulfite oxidase uses a slightly different cofactor sulfuration and is preserved).
In contrast to molybdenum cofactor deficiency, both xanthinuria types are generally benign. The defining biochemical findings are very low serum and urinary uric acid (often <1 mg/dL) and markedly elevated urinary xanthine. The major clinical risk is xanthine urolithiasis — xanthine is less soluble than uric acid, and accumulation in the urine can precipitate to form kidney stones. Recommended management is high fluid intake, dietary purine restriction, and occasional urine alkalinization. Some patients are asymptomatic and detected only incidentally on biochemistry.
Xanthinuria type II patients additionally have impaired metabolism of aldehyde-oxidase substrate drugs (zaleplon, famciclovir, methotrexate, etc.) — relevant when these medications are prescribed.
Molybdenum, Copper, and Gout Risk
A nuanced question is whether dietary molybdenum status influences gout risk. The biochemistry is clear: xanthine oxidase requires the molybdopterin cofactor, so adequate molybdenum is necessary for normal uric acid production. Severe molybdenum deficiency (in the TPN setting) produces hypouricemia, not hyperuricemia.
Conversely, very high molybdenum intake (>500 mcg/day chronically, well above the 45 mcg RDA) has been associated with elevated uric acid in occupational exposure studies from molybdenum mining and processing regions (Armenia, parts of the Soviet Union historical literature). Whether dietary or supplemental molybdenum at typical supplement doses (50–500 mcg/day) materially affects gout risk is unclear — most clinical reports do not show meaningful uric acid elevation at these doses.
For gout patients considering molybdenum supplementation (for sulfite metabolism or other reasons), the prudent approach is:
- Keep intake at or near the 45 mcg RDA unless there is a specific clinical reason for higher dosing
- Monitor serum uric acid before and 6–12 weeks after supplement initiation
- Avoid high-dose molybdenum (>500 mcg/day) without specific indication
- Maintain adequate copper intake to balance the molybdenum-copper antagonism
The copper-molybdenum-sulfur triangle is particularly relevant in agricultural contexts (where ruminant nutrition has long balanced these three minerals) and in Wilson disease treatment (where ammonium tetrathiomolybdate is used to decopper patients). For human dietary supplementation, the practical implication is that high molybdenum can drive functional copper deficiency, which has its own constellation of problems (anemia, neutropenia, neurological symptoms).
Tumor Lysis Syndrome and Ischemia-Reperfusion
Xanthine oxidase plays a central role in two acute medical emergencies:
Tumor lysis syndrome (TLS) occurs when chemotherapy or radiation triggers massive cancer cell death, releasing intracellular contents including purines, potassium, and phosphate. The purine load floods through xanthine oxidase, generating enormous amounts of uric acid that exceed renal excretory capacity, precipitate as uric acid crystals in renal tubules, and cause acute kidney injury. The phosphate load drives hyperphosphatemia and hypocalcemia. The potassium release causes hyperkalemia. The combined effects can be rapidly fatal.
Prophylaxis for high-risk patients (high tumor burden lymphoma, leukemia, high-grade solid tumors) involves:
- Allopurinol initiated 1–2 days before chemotherapy to block xanthine oxidase
- Rasburicase (recombinant urate oxidase) for high-risk cases — converts existing uric acid to soluble allantoin, providing rapid urate reduction
- Aggressive hydration
- Urine alkalinization (now somewhat controversial)
- Frequent electrolyte and renal function monitoring
Ischemia-reperfusion injury is the classic XO-mediated injury pattern discussed above. Allopurinol pretreatment in elective surgery (cardiac, transplant) and in ischemic stroke has been studied as a strategy to reduce reperfusion injury. Results are mixed but generally favorable; the practical adoption varies by institution.
Key Research Papers
- Harrison R (2004). Physiological roles of xanthine oxidoreductase. Drug Metabolism Reviews. — PubMed
- Choi HK et al. (2004). Purine-rich foods, dairy and protein intake, and the risk of gout in men. NEJM. — PubMed
- White WB et al. (2018). Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout (CARES trial). NEJM. — PubMed
- Becker MA et al. (2005). Febuxostat compared with allopurinol in patients with hyperuricemia and gout. NEJM. — PubMed
- FitzGerald JD et al. (2020). 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis & Rheumatology. — PubMed
- Hung SI et al. (2005). HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. PNAS. — PubMed
- Ames BN et al. (1981). Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer. PNAS. — PubMed
- Hediger MA et al. (2005). Molecular physiology of urate transport. Physiology. — PubMed
- Granger DN (1988). Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. American Journal of Physiology. — PubMed
- Cardoso AS et al. (2014). Xanthine oxidase and oxidative stress in the management of cardiovascular and renal disease. Current Hypertension Reports. — PubMed
- Coiffier B et al. (2008). Guidelines for the management of pediatric and adult tumor lysis syndrome. Journal of Clinical Oncology. — PubMed
- Choi HK, Curhan G (2008). Soft drinks, fructose consumption, and the risk of gout in men. BMJ. — PubMed
PubMed Topic Searches
- PubMed: Xanthine oxidase uric acid
- PubMed: Gout pathophysiology treatment
- PubMed: Allopurinol / febuxostat / gout
- PubMed: Hereditary xanthinuria
- PubMed: Uric acid antioxidant peroxynitrite
Connections
- Molybdenum Benefits Hub
- Molybdenum Overview
- Molybdenum for Detoxification
- Molybdenum for Sulfite Metabolism
- Molybdenum for Iron Utilization
- Gout
- Uric Acid Test
- Kidney Stones
- Kidney Disease
- Heart Failure
- Hypertension
- Metabolic Syndrome
- Copper
- Organ Meats (Purines)
- High-Fructose Corn Syrup
- Vitamin C (Uricosuric)
- Oxidative Stress
- All Minerals