Molybdenum for Iron Utilization
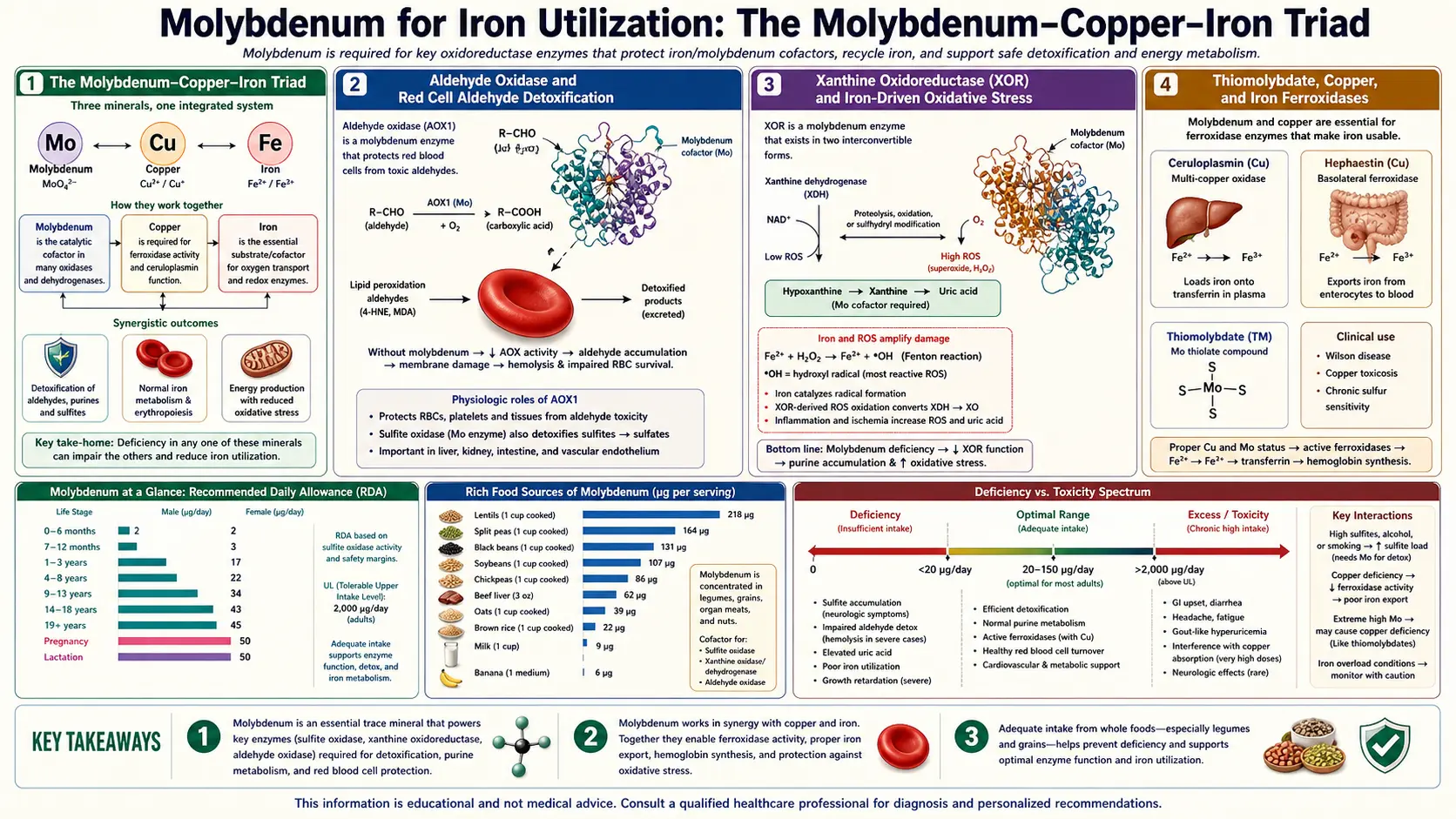
The relationship between molybdenum and iron metabolism is more intricate than the textbook accounts suggest. Molybdenum's direct role is through aldehyde oxidase, which oxidizes biogenic aldehydes that can otherwise damage red blood cells, and through xanthine oxidoreductase, which sits at a key crossroads of iron-dependent oxidative chemistry. But the larger story is the molybdenum-copper-iron triad — three trace minerals whose biological availability is mechanically linked: high molybdenum drives copper deficiency via thiomolybdate complex formation; copper deficiency in turn impairs iron utilization through ceruloplasmin and hephaestin (the copper-dependent ferroxidases that load iron onto transferrin); and the resulting "low-copper anemia" is biochemically indistinguishable from iron-deficiency anemia until copper is supplemented. This deep-dive maps the molecular mechanisms, the rare TPN-driven anemia case reports that revealed the molybdenum-iron connection, the diagnostic implications, and the cofactor biosynthesis defects (MoCoSulfurase deficiency) that disrupt the system.
Table of Contents
- The Molybdenum-Copper-Iron Triad
- Aldehyde Oxidase and Red Cell Aldehyde Detoxification
- Xanthine Oxidoreductase and Iron-Driven Oxidative Stress
- Thiomolybdate, Copper, and Iron Ferroxidases
- TPN-Driven Molybdenum Deficiency and Anemia
- MoCo Biosynthesis Defects (MOCS1/2/3, MOCOS, GPHN)
- Molybdenum Cofactor Sulfurase Deficiency
- Iron Overload, Copper, and the Molybdenum Question
- Diagnostic Implications — When to Think Molybdenum
- Supplementation: Balancing the Three Minerals
- Key Research Papers
- Connections
- Featured Videos
The Molybdenum-Copper-Iron Triad
Three transition metals — molybdenum (Mo), copper (Cu), and iron (Fe) — have biochemically intertwined fates. Each is required in trace amounts, each cycles between oxidation states to perform electron-transfer catalysis, and each can substitute for or block the others under specific conditions. The mechanical links that matter for iron utilization:
- Mo ↔ Cu antagonism — molybdate (MoO42−) in the presence of sulfide (HS−) forms thiomolybdate complexes (MoOS32−, MoS42−) that bind copper with very high affinity. High dietary molybdenum (especially with high sulfur) can therefore deplete the copper pool, producing functional copper deficiency even when copper intake is adequate. This effect is dramatically amplified in ruminant animals (where rumen sulfate-reducing bacteria generate abundant sulfide) and is the basis for "molybdenum toxicity" in cattle grazing on high-Mo pastures.
- Cu → Fe ferroxidase function — copper is the essential catalytic metal in ceruloplasmin (the plasma ferroxidase) and hephaestin (the membrane-bound enterocyte ferroxidase). These enzymes oxidize ferrous iron (Fe2+) to ferric iron (Fe3+), the form that transferrin binds and circulates. Without functional ferroxidase, iron cannot be loaded onto transferrin, cannot be transported to bone marrow erythroid precursors, and cannot be incorporated into hemoglobin — even when iron stores are adequate.
- Net effect — high molybdenum → depleted copper → impaired ferroxidase activity → iron "trapped" in stores but unavailable for erythropoiesis → "low-copper anemia" that is biochemically a hypochromic microcytic anemia indistinguishable from iron-deficiency anemia at first glance, with normal or even elevated serum ferritin (iron stores are adequate) and low serum copper / low ceruloplasmin (the actual rate-limiting deficiency).
The clinical signature of low-copper anemia: hypochromic microcytic red blood cells, often with concurrent neutropenia (copper is required for neutrophil maturation), normal or elevated ferritin, low serum iron, low transferrin saturation, low serum copper, low ceruloplasmin, sometimes high free Mo. Iron supplementation does not correct the anemia — in fact it may worsen it by saturating ferroxidase capacity. Copper supplementation rapidly corrects the anemia within weeks.
This is why molybdenum supplementation must always be considered in the context of copper status, and why "iron-refractory anemia" should prompt consideration of copper (and indirectly molybdenum) status before further iron loading.
Aldehyde Oxidase and Red Cell Aldehyde Detoxification
Aldehyde oxidase (AOX1) — one of the three classical molybdenum-dependent enzymes — oxidizes a broad range of aldehydes, including biogenic aldehydes generated during normal cellular metabolism. Several of these AOX1 substrates are particularly relevant to red blood cell biology:
- Pyridoxal (vitamin B6 aldehyde) — AOX1 converts pyridoxal to pyridoxic acid. Pyridoxal-5-phosphate (the active B6 form) is essential for hemoglobin synthesis: the rate-limiting enzyme of heme synthesis, ALA synthase, requires PLP as cofactor. Disturbance of B6 metabolism can therefore impair hemoglobin production — one of the rarer causes of sideroblastic anemia.
- Retinaldehyde — AOX1 contributes to converting retinaldehyde to retinoic acid, which is required for normal erythropoiesis through retinoic acid receptor signaling in the bone marrow erythroid niche. Vitamin A deficiency is a recognized cause of anemia that is often refractory to iron supplementation.
- 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA) — lipid peroxidation products. Red blood cells, with their high membrane lipid content and oxygen exposure, generate these continuously. AOX1 in the liver clears the circulating pool; reduced AOX1 activity allows accumulation that can damage red cell membranes and shorten erythrocyte lifespan.
- Biogenic aldehydes from neurotransmitter and amine metabolism — including the aldehyde intermediates of dopamine, serotonin, and various dietary amines
The clinical signal of severe AOX1 dysfunction (combined molybdenum cofactor deficiency, MoCoSulfurase deficiency, or rare AOX1 polymorphisms) includes accumulation of aldehyde metabolites that can be measured in urine, and in some case reports, hemolytic features and reduced erythrocyte lifespan.
Xanthine Oxidoreductase and Iron-Driven Oxidative Stress
Xanthine oxidase (XO) and xanthine dehydrogenase (XDH) sit at a key intersection of iron-dependent oxidative chemistry. The XO form generates superoxide (O2−) and hydrogen peroxide (H2O2) as catalytic byproducts. In the presence of free iron, hydrogen peroxide drives the Fenton reaction:
H2O2 + Fe2+ → ·OH + OH− + Fe3+
Hydroxyl radical (·OH) is the most reactive species in biological chemistry, indiscriminately damaging lipids, proteins, and DNA. Fenton-driven oxidative damage is a central mechanism in:
- Iron-overload toxicity — in hereditary hemochromatosis, transfusion-dependent thalassemia, and other iron-overload states
- Ischemia-reperfusion injury — XO-derived H2O2 plus released iron from damaged ferritin creates a particularly toxic combination
- Atherosclerosis and vascular disease — vascular wall XO plus circulating iron contribute to LDL oxidation
The therapeutic implication is that xanthine oxidase inhibition (with allopurinol or febuxostat) reduces H2O2 generation and therefore reduces Fenton-driven damage in the presence of iron overload. Several small studies have explored allopurinol as adjunctive therapy in conditions where iron-driven oxidative damage matters (thalassemia, sickle cell disease, ischemia-reperfusion in cardiac surgery), with generally favorable but not definitive results.
The molybdenum implication: adequate molybdenum supports normal XO function; severe deficiency abolishes XO activity (and therefore both purine catabolism and H2O2 generation). The XDH form — the dominant physiological form — produces much less ROS than the XO form, so the net effect of normal molybdenum status is primarily metabolic rather than oxidative.
Thiomolybdate, Copper, and Iron Ferroxidases
The mechanism by which excess molybdenum impairs iron utilization runs through copper. In the gut and in plasma, molybdate ions can react with sulfide (HS−, generated by sulfate-reducing bacteria in the gut and by endogenous cysteine catabolism) to form thiomolybdates:
MoO42− + HS− → MoO3S2− + OH−
MoO42− + 2 HS− → MoO2S22− + 2 OH−
MoO42− + 3 HS− → MoOS32− + 3 OH−
MoO42− + 4 HS− → MoS42− (tetrathiomolybdate) + 4 OH−
The fully thio-substituted form, tetrathiomolybdate (TM), is the most potent copper chelator in the series and binds copper with picomolar affinity, forming a stable Mo-Cu-S cluster that the body cannot easily disassemble. The complex is then excreted via the bile.
This mechanism is therapeutically exploited as ammonium tetrathiomolybdate in Wilson disease, where it is used to remove tissue copper. The same mechanism is the basis for the agricultural problem of molybdenum-induced copper deficiency in ruminants grazing on high-Mo pastures.
The iron consequence:
- Ceruloplasmin requires 6 copper atoms per molecule for its ferroxidase activity. Copper depletion produces apo-ceruloplasmin (the copper-free form) which has no ferroxidase function. Plasma ferrous iron cannot be oxidized to ferric iron and therefore cannot bind transferrin.
- Hephaestin, the membrane-bound ferroxidase at the basolateral surface of enterocytes, is also copper-dependent. Without hephaestin, dietary iron absorbed through the apical DMT1 transporter cannot exit the enterocyte through ferroportin in a usable form.
- The net effect is functional iron deficiency despite adequate iron intake and stores
This sequence — high Mo → thiomolybdate → Cu sequestration → ferroxidase failure → iron unavailability — explains why molybdenum status matters for iron utilization indirectly through copper.
TPN-Driven Molybdenum Deficiency and Anemia
Direct molybdenum deficiency in humans has been documented essentially only in patients on long-term total parenteral nutrition (TPN) without molybdenum supplementation. The landmark case is Abumrad et al. 1981 (Am J Clin Nutr): a 24-year-old patient with Crohn's disease on prolonged TPN developed:
- Tachycardia and tachypnea
- Mental status changes progressing to coma
- Visual scotomata
- Sulfite oxidase failure (high urinary sulfite, low urinary sulfate, low serum methionine due to methionine catabolism inhibition by sulfite accumulation)
- Xanthine oxidase failure (low serum and urinary uric acid, elevated urinary xanthine and hypoxanthine)
- Resolution within days of adding ammonium molybdate to the TPN formula
The case did not specifically emphasize iron-utilization anemia (the patient's primary problem was the metabolic encephalopathy), but several subsequent TPN molybdenum-deficiency reports have noted concurrent anemia features and impaired response to iron supplementation, consistent with the thiomolybdate-copper mechanism. Modern TPN formulations include molybdenum routinely (typically as ammonium molybdate, 20–200 mcg/day depending on patient population and ASPEN guidelines), and the Abumrad-type presentation is no longer seen in well-managed TPN programs.
Infant TPN was historically a higher-risk setting because pediatric trace mineral formulations have evolved over time. Several case reports from the 1980s described TPN-fed premature infants with molybdenum deficiency presenting with refractory anemia, sulfite toxicity, and developmental impairment that responded to molybdenum supplementation.
MoCo Biosynthesis Defects (MOCS1/2/3, MOCOS, GPHN)
The biosynthesis of the molybdopterin cofactor (Moco) requires the coordinated function of multiple gene products:
- MOCS1 — converts GTP to cyclic pyranopterin monophosphate (cPMP), the first committed step
- MOCS2 — converts cPMP to molybdopterin
- MOCS3 — provides sulfur transfer for the molybdopterin synthesis
- GPHN (gephyrin) — inserts molybdenum into the molybdopterin scaffold
- MOCOS (molybdenum cofactor sulfurase) — performs the terminal sulfuration step required for xanthine oxidase and aldehyde oxidase function specifically (sulfite oxidase is preserved)
Mutations in MOCS1, MOCS2, MOCS3, or GPHN produce molybdenum cofactor deficiency (MoCD), with simultaneous failure of all three molybdoenzymes. The clinical presentation is dominated by sulfite toxicity (see the Sulfite Metabolism page for full details), but the concurrent loss of xanthine and aldehyde oxidase activity has documented effects on iron metabolism:
- Accumulation of aldehyde substrates that can damage red blood cell membranes
- Loss of pyridoxal → pyridoxic acid oxidation, potentially affecting B6 status and hemoglobin synthesis
- Accumulation of xanthine, which can substitute partially for hypoxanthine in salvage reactions but accumulates in tissues and can precipitate as crystals in kidneys
- Some case reports describe mild hemolytic features in MoCD patients
MOCS1 deficiency (MoCD type A) is the one form for which a replacement therapy exists: fosdenopterin (cPMP, brand name Nulibry) — a synthetic version of the MOCS1 product — was FDA-approved in February 2021. Daily intravenous infusion initiated within days of birth restores cofactor biosynthesis and rescues all three enzyme activities. Treatment is lifelong.
Molybdenum Cofactor Sulfurase Deficiency
A particularly interesting variant is molybdenum cofactor sulfurase (MOCOS) deficiency, also known as xanthinuria type II. The MOCOS enzyme performs the final sulfuration step that activates xanthine oxidase and aldehyde oxidase (but not sulfite oxidase, which uses a separate sulfuration mechanism via SUOX itself).
MOCOS-deficient patients therefore have:
- Absent xanthine oxidase activity (presenting as xanthinuria — very low uric acid, elevated urinary xanthine, risk of xanthine kidney stones)
- Absent aldehyde oxidase activity (impaired metabolism of AOX1 substrate drugs)
- Preserved sulfite oxidase activity (and therefore none of the catastrophic neurological features of MoCD or ISOD)
The clinical phenotype is generally mild — xanthine kidney stones, low serum uric acid, and clinically silent unless drug metabolism issues arise. Some patients are detected only incidentally on biochemistry showing low uric acid.
The relevance to iron utilization: MOCOS deficiency affects the AOX1-mediated aldehyde clearance pathway discussed above. Whether this contributes to anemia or impaired iron utilization in MOCOS-deficient patients is poorly characterized in the literature — the cohort is small and clinical reports focus on the more prominent xanthinuria features.
Iron Overload, Copper, and the Molybdenum Question
The reverse problem — iron overload — is far more common than iron deficiency in adult Western populations, particularly among men, postmenopausal women, and those with hereditary hemochromatosis (HFE C282Y/H63D variants). Iron overload drives Fenton-mediated oxidative damage, contributes to liver fibrosis, cardiomyopathy, diabetes, and hypogonadism, and is the focus of much clinical attention.
The molybdenum-copper-iron relationship in iron overload:
- Adequate ceruloplasmin (and therefore adequate copper) is essential for maintaining the ferric-bound transferrin pool. In aceruloplasminemia (genetic absence of ceruloplasmin), iron accumulates pathologically in tissues despite normal HFE genotype — an iron-loading anemia caused by inability to mobilize iron stores
- High-dose molybdenum supplementation that depletes copper could in principle worsen iron loading or impair iron mobilization in patients with subclinical ceruloplasmin insufficiency
- The tetrathiomolybdate / Wilson disease parallel is instructive: TM is used to chelate copper in Wilson disease (where copper overload is the problem); the same chemistry inadvertently happens in livestock with high Mo and high S intake, producing copper-deficient cattle
For practical purposes, individuals with iron overload (hereditary hemochromatosis, thalassemia, transfusion-dependent anemias) should not pursue aggressive molybdenum supplementation unless there is a specific clinical reason. The 45 mcg/day RDA met through diet is generally sufficient and does not perturb the iron-copper-molybdenum balance.
Conversely, Morley Robbins's work on copper-iron dysregulation emphasizes that "iron-deficiency anemia" is often actually copper-deficiency anemia in disguise, and that treating with high-dose iron without addressing copper status can worsen long-term outcomes by driving iron accumulation in tissues without functional transport.
Diagnostic Implications — When to Think Molybdenum
Molybdenum should be considered in the workup of iron-related issues in specific clinical scenarios:
- Iron-refractory anemia — hypochromic microcytic anemia that does not respond to iron supplementation. Check serum copper and ceruloplasmin; if low, consider both copper deficiency primary and the molybdenum-thiomolybdate-copper axis. Inquire about molybdenum-rich supplements (a few "all in one" trace mineral products contain disproportionately high molybdenum that can sequester copper)
- Long-term TPN — verify the parenteral formulation includes both molybdenum and copper at appropriate doses (ASPEN guidelines)
- Inflammatory bowel disease with persistent anemia — multi-mineral status assessment including iron studies, B12, folate, copper, ceruloplasmin, and consideration of molybdenum, particularly if the patient is on supplements containing molybdenum
- Bariatric surgery — gastric bypass and duodenal switch dramatically alter trace mineral absorption. Copper deficiency is recognized after bariatric surgery; molybdenum status is less well-studied but warrants attention in difficult cases
- Wilson disease patients on tetrathiomolybdate — close iron monitoring is required because the therapy specifically depletes the ferroxidase machinery
- Cystic fibrosis patients — pancreatic insufficiency-driven fat-soluble vitamin malabsorption may be accompanied by trace mineral status disturbances
- Patients with unexplained xanthinuria — low uric acid (<1 mg/dL) without obvious cause should prompt consideration of xanthine oxidase deficiency (XDH variants) or MOCOS deficiency, both of which have implications for AOX1-substrate drug metabolism
Laboratory testing for molybdenum status is not routinely available in most clinical labs. Serum molybdenum measurement is possible at specialty labs but interpretation is difficult; urinary sulfite and xanthine measurement provides indirect evidence of molybdoenzyme activity; serum uric acid below 2 mg/dL without identifiable cause is a red flag for xanthine oxidase dysfunction.
Supplementation: Balancing the Three Minerals
For practical supplementation guidance when iron utilization is a concern:
- Molybdenum — meet the 45 mcg/day RDA from food (legumes, whole grains, nuts) without routine supplementation unless there is a specific indication. If supplementing for sulfite-metabolism or detoxification purposes, stay at 75–500 mcg/day and monitor copper status
- Copper — meet the 900 mcg/day RDA from food (organ meats, shellfish, cocoa, sesame seeds, sunflower seeds) or low-dose supplement (1–2 mg/day). Avoid simultaneous high-zinc supplementation, which can drive copper deficiency via metallothionein induction in enterocytes
- Iron — meet from food (red meat, poultry, fish, beans, dark leafy greens with vitamin C) unless there is documented deficiency. Iron supplements should be reserved for documented deficiency with appropriate workup; routine high-dose iron in postmenopausal women and men risks iron loading
- Zinc — relevant because of zinc-copper antagonism. Long-term high-dose zinc (>50 mg/day) without copper repletion is a recognized cause of copper deficiency and secondary anemia
- Vitamin C — supports non-heme iron absorption; also has uricosuric effect (mildly lowers serum uric acid)
- Vitamin A — required for normal erythropoiesis; deficiency contributes to anemia that may be refractory to iron alone
The most common practical scenario is the patient on a multi-mineral supplement that contains disproportionately high molybdenum and low copper, who develops mild copper deficiency and "iron-refractory anemia" over months. Reading the supplement label carefully, adjusting the mineral balance, and rechecking after 8–12 weeks often resolves the problem without elaborate workup.
Key Research Papers
- Abumrad NN et al. (1981). Amino acid intolerance during prolonged total parenteral nutrition reversed by molybdate therapy. American Journal of Clinical Nutrition. — PubMed
- Suttle NF (2010). Mineral Nutrition of Livestock (chapter on Cu-Mo-S interactions). CABI. — PubMed
- Brewer GJ et al. (2006). Tetrathiomolybdate for the treatment of Wilson disease. Archives of Neurology. — PubMed
- Hellman NE, Gitlin JD (2002). Ceruloplasmin metabolism and function. Annual Review of Nutrition. — PubMed
- Halfdanarson TR et al. (2008). Hematological manifestations of copper deficiency: a retrospective review. European Journal of Haematology. — PubMed
- Williams DM (1983). Copper deficiency in humans. Seminars in Hematology. — PubMed
- Anderson GJ, Frazer DM (2017). Current understanding of iron homeostasis. American Journal of Clinical Nutrition. — PubMed
- Schwarz G, Mendel RR (2006). Molybdenum cofactor biosynthesis and molybdenum enzymes. Annual Review of Plant Biology. — PubMed
- Ichida K et al. (2012). Identification of MOCOS gene mutations in a patient with xanthinuria type II. Human Molecular Genetics. — PubMed
- Vyoral D, Petrak J (2017). Therapeutic potential of hepcidin — the master regulator of iron metabolism. Pharmacological Research. — PubMed
- Mills KC, Curry SC (1994). Acute iron poisoning. Emergency Medicine Clinics of North America. — PubMed
- Pietrangelo A (2010). Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. — PubMed
PubMed Topic Searches
- PubMed: Molybdenum-copper-iron interaction
- PubMed: Ceruloplasmin ferroxidase iron
- PubMed: Copper deficiency anemia
- PubMed: Tetrathiomolybdate Wilson disease
- PubMed: Fosdenopterin / cPMP therapy
Connections
- Molybdenum Benefits Hub
- Molybdenum Overview
- Molybdenum for Detoxification
- Molybdenum for Sulfite Metabolism
- Molybdenum for Uric Acid
- Copper
- Zinc
- Sulfur
- Copper-Iron Dysregulation
- Whole-Food Copper Sources
- Anemia
- Vitamin C (Iron Absorption)
- Vitamin A (Erythropoiesis)
- Vitamin B6 (Hemoglobin Synthesis)
- Vitamin B12
- Liver Disease
- Organ Meats
- All Minerals