Vasculitis
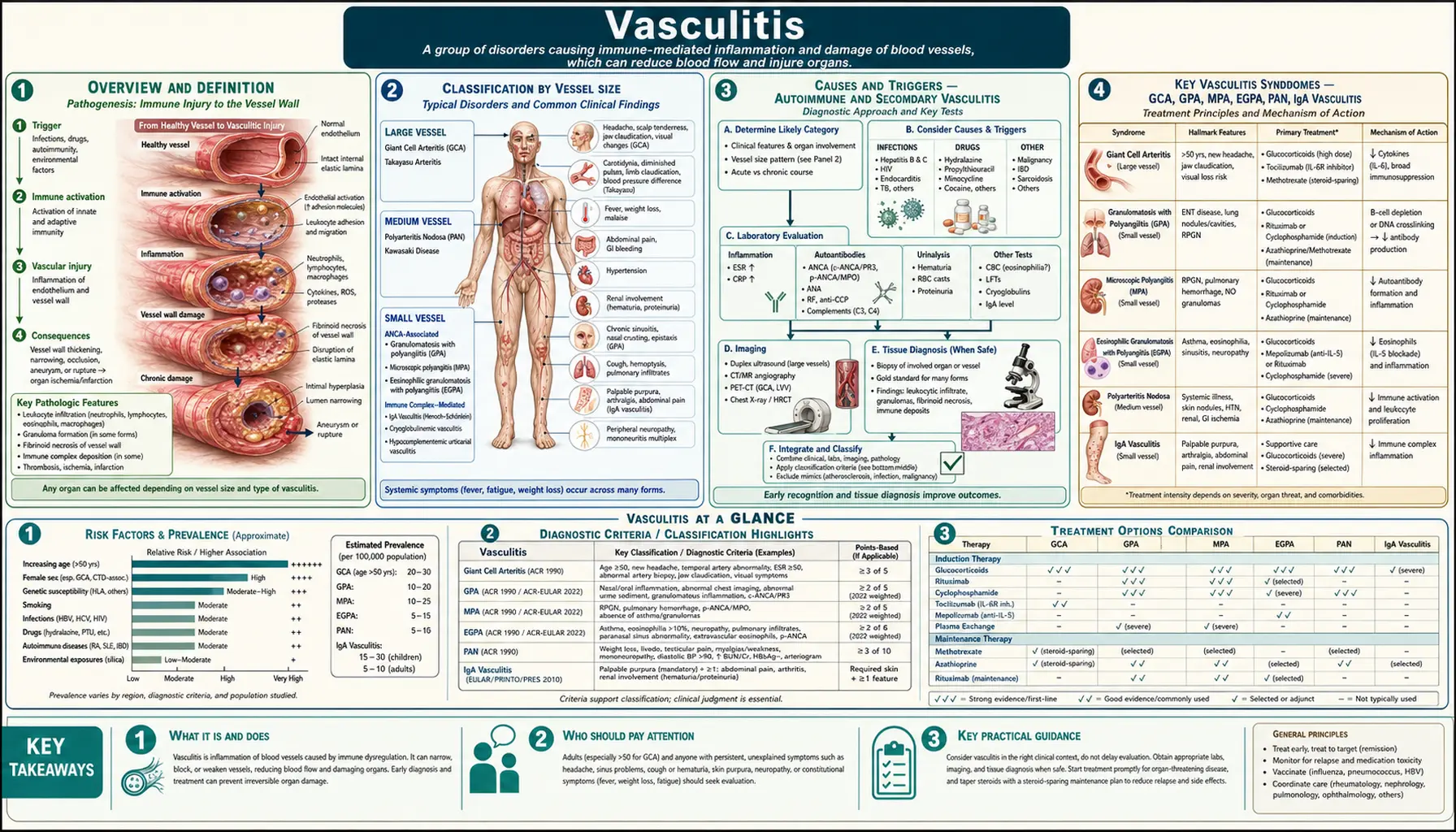
Table of Contents
- Overview and Definition
- Classification by Vessel Size
- Causes and Triggers — Autoimmune and Secondary Vasculitis
- Key Vasculitis Syndromes — GCA, GPA, MPA, EGPA, PAN, IgA Vasculitis
- Symptoms and Organ Involvement
- Diagnosis — Biopsy, ANCA Testing, Imaging
- Treatment — Corticosteroids, Immunosuppressants, Biologics
- Natural and Nutritional Approaches
- Disease Course and Prognosis
- Complications and Organ Damage
- Prevention and Monitoring
- Key Research Papers
- Connections
- Featured Videos
Overview and Definition
Vasculitis is a group of disorders characterized by inflammation of blood vessel walls — arteries, veins, or capillaries. When vessel walls become inflamed, they can thicken, weaken, narrow, or scar, restricting blood flow to tissues and organs. The result ranges from mild skin rashes to life-threatening organ failure, depending on which vessels are involved and how aggressively the disease progresses.
Vasculitis is not a single disease but a family of more than 20 distinct conditions, each defined by the caliber of vessel affected, the presence or absence of specific antibodies (particularly ANCA — antineutrophil cytoplasmic antibodies), and the pattern of organ involvement. Some forms are primary (driven by the immune system attacking vessels directly), while others are secondary to infections, medications, malignancy, or other autoimmune diseases such as lupus or rheumatoid arthritis.
Globally, vasculitis affects roughly 150–200 people per million per year across all forms combined, though estimates vary widely by form and geography. Giant Cell Arteritis (GCA) is the most common primary vasculitis in adults over 50 in the Western world, while IgA Vasculitis (Henoch-Schönlein Purpura) is the most common form in children. ANCA-associated vasculitides (GPA, MPA, EGPA) together affect approximately 50 per million per year in Europe and North America.
Early diagnosis is critical. GCA can cause permanent vision loss within days if untreated. ANCA-associated vasculitis can destroy kidney function within weeks. Recognition of the characteristic symptom clusters — unexplained fever with weight loss, rash, hematuria, hemoptysis, sinus destruction, or new-onset headache in an older adult — is the key to preventing irreversible damage.
Classification by Vessel Size
The 2012 revised Chapel Hill Consensus Conference (CHCC) Nomenclature provides the internationally accepted framework for classifying vasculitis by the predominant vessel size affected. This classification guides diagnosis, treatment selection, and prognosis.
Large Vessel Vasculitis
Involves the aorta and its major branches. The two primary forms are:
- Giant Cell Arteritis (GCA / Temporal Arteritis): The most common large-vessel vasculitis, almost exclusively affecting people over 50. Involves the cranial branches of the aorta (temporal, ophthalmic, posterior ciliary arteries) and the aorta itself. Hallmark: granulomatous inflammation with multinucleated giant cells. Risk of blindness from anterior ischemic optic neuropathy is up to 20% if untreated.
- Takayasu Arteritis: A granulomatous vasculitis of the aorta and its major branches, predominantly affecting young women (under 40) of Asian descent. Causes stenosis, occlusion, or aneurysm of the subclavian, carotid, renal, and mesenteric arteries. Presents with limb claudication, absent pulses, and hypertension from renal artery involvement.
Medium Vessel Vasculitis
Involves the main visceral arteries and their branches, including renal, hepatic, coronary, and mesenteric arteries.
- Polyarteritis Nodosa (PAN): Necrotizing arteritis of medium and small arteries without glomerulonephritis or capillaritis. Strongly associated with hepatitis B infection. Presents with fever, weight loss, livedo reticularis, mononeuritis multiplex, and renal infarction (hypertension but not hematuria, distinguishing it from ANCA vasculitis).
- Kawasaki Disease: Primarily affects children under 5. Necrotizing arteritis involving the coronary arteries, with risk of coronary artery aneurysm. Presents with prolonged fever, rash, conjunctivitis, strawberry tongue, and desquamation of the fingertips. Treated with IVIG and aspirin.
Small Vessel Vasculitis — ANCA-Associated
These three forms are grouped together by their association with antineutrophil cytoplasmic antibodies (ANCA) and their tendency to cause necrotizing inflammation of small vessels, particularly glomerulonephritis and pulmonary capillaritis.
- Granulomatosis with Polyangiitis (GPA, formerly Wegener's Granulomatosis): Granulomatous inflammation of the upper and lower respiratory tracts plus necrotizing glomerulonephritis. Associated with PR3-ANCA (c-ANCA) in ~90% of generalized disease. Classic triad: destructive sinusitis/ear disease, pulmonary infiltrates or nodules, and renal involvement.
- Microscopic Polyangiitis (MPA): Necrotizing vasculitis without granulomatous inflammation. Associated with MPO-ANCA (p-ANCA) in ~60–70%. Causes rapidly progressive glomerulonephritis and pulmonary hemorrhage (pulmonary-renal syndrome), but lacks the upper respiratory tract destruction seen in GPA.
- Eosinophilic Granulomatosis with Polyangiitis (EGPA, formerly Churg-Strauss Syndrome): Granulomatous inflammation rich in eosinophils, typically in the setting of asthma and peripheral blood eosinophilia (>10%). MPO-ANCA present in ~40%. Often involves the heart (eosinophilic myocarditis), peripheral nerves, and skin. Associated with leukotriene receptor antagonist use in rare cases.
Small Vessel Vasculitis — Immune Complex-Mediated
- IgA Vasculitis (Henoch-Schönlein Purpura, HSP): IgA immune complex deposition in small vessel walls. Most common vasculitis in children, often triggered by upper respiratory infections (Group A streptococcus). Tetrad: palpable purpura (buttocks/legs), arthritis, abdominal pain, and renal involvement (IgA nephropathy pattern).
- Cryoglobulinemic Vasculitis: Caused by immune complexes containing cryoglobulins (immunoglobulins that precipitate in cold). Strongly associated with hepatitis C infection (>80% of mixed cryoglobulinemia). Presents with purpura, weakness, arthralgias, and peripheral neuropathy. Antiviral therapy for HCV is the cornerstone of treatment.
- Hypersensitivity Vasculitis (Leukocytoclastic Vasculitis): Post-exposure to drugs, infections, or food antigens; predominantly skin-limited; resolves with removal of the trigger.
Causes and Triggers — Autoimmune and Secondary Vasculitis
The etiology of vasculitis is incompletely understood but involves a convergence of genetic susceptibility, immune dysregulation, and environmental or infectious triggers that break normal vascular tolerance.
Primary (Idiopathic) Autoimmune Mechanisms
In ANCA-associated vasculitis, the current model holds that primed neutrophils express PR3 or MPO on their surface. ANCA antibodies bind these antigens, activating the neutrophils to release toxic granule contents and reactive oxygen species directly into vessel walls. This neutrophil-endothelial interaction drives the necrotizing inflammation that destroys capillary walls. Why ANCA develop in the first place remains unclear, but genetic factors (HLA-DP for GPA, HLA-DQ for MPA), silica dust exposure, and prior infections (particularly Staphylococcus aureus nasal carriage, which promotes PR3 expression) are implicated.
In GCA, activated CD4+ T cells infiltrate the arterial wall, differentiating into Th1 and Th17 subsets. Interleukin-6 (IL-6) is a key cytokine driver — explaining why tocilizumab (an IL-6 receptor blocker) is effective. Dendritic cells in the adventitia act as the initial antigen-presenting sentinels, though the triggering antigen has not been identified.
Infectious Triggers
- Hepatitis B: Immune complex deposition triggers classic Polyarteritis Nodosa. Antiviral therapy is curative when started early.
- Hepatitis C: Drives mixed cryoglobulinemia and its associated vasculitis; direct-acting antivirals (DAAs) have transformed outcomes.
- Group A Streptococcus: Post-streptococcal IgA deposition triggers IgA Vasculitis (HSP) in children; most recover fully within 4–6 weeks.
- Staphylococcus aureus: Nasal carriage associated with GPA relapse, possibly through molecular mimicry with PR3.
- HIV, CMV, Parvovirus B19: Associated with secondary vasculitis presentations.
Drug-Induced Vasculitis
Multiple drugs can trigger ANCA-positive vasculitis, most notably propylthiouracil, hydralazine, minocycline, levamisole-adulterated cocaine, and allopurinol. Drug-induced ANCA vasculitis often produces both MPO and PR3 antibodies simultaneously — a pattern rare in primary disease — and typically remits with drug discontinuation, though immunosuppression is sometimes needed for severe organ involvement.
Malignancy-Associated Vasculitis
Lymphomas and solid tumors can provoke paraneoplastic vasculitis. Hairy cell leukemia is classically associated with PAN-like presentations. Screening for underlying malignancy is warranted in older adults with newly diagnosed vasculitis, especially when constitutional symptoms are prominent.
Genetic and Environmental Risk Factors
HLA-DPB1*04:01 is strongly associated with GPA (PR3-ANCA vasculitis). Occupational silica and solvent exposure increases GPA risk two- to fourfold. Smoking is associated with EGPA and with worse outcomes across forms. Geographic clustering of GCA in Scandinavian populations and their descendants suggests both genetic and possibly shared environmental (viral?) factors.
Key Vasculitis Syndromes — GCA, GPA, MPA, EGPA, PAN, IgA Vasculitis
Giant Cell Arteritis (GCA)
GCA is an ophthalmologic emergency. The typical patient is a woman over 70 of Northern European descent presenting with new-onset severe temporal headache, scalp tenderness, jaw claudication (pain with chewing), and systemic symptoms (fever, night sweats, weight loss). Erythrocyte sedimentation rate (ESR) is typically >50 mm/hr (often >100), and C-reactive protein (CRP) is elevated. Diagnosis is confirmed by temporal artery biopsy showing granulomatous inflammation with giant cells — though sensitivity is ~85% because of skip lesions, and clinical diagnosis is acceptable when biopsy is negative with high pre-test probability. Large-vessel GCA (aortic involvement) occurs in 20–40% of patients and can cause aortic aneurysm years after the cranial disease resolves. The GiACTA trial established tocilizumab (162 mg weekly subcutaneous) as a steroid-sparing agent, achieving sustained remission in 56% vs. 14% with prednisone alone.
Granulomatosis with Polyangiitis (GPA)
GPA classically destroys the upper respiratory tract — causing chronic sinusitis that fails antibiotics, bloody nasal discharge, nasal septal perforation, and the characteristic saddle-nose deformity from cartilage collapse. Subglottic stenosis can develop insidiously. Pulmonary involvement causes hemoptysis, cavitating nodules, and diffuse alveolar hemorrhage. Renal involvement (rapidly progressive glomerulonephritis with red cell casts) can progress to dialysis-dependent kidney failure within weeks. Orbital pseudotumor causes proptosis. PR3-ANCA (c-ANCA pattern) is positive in ~90% of active generalized GPA and serves as both a diagnostic and disease-activity marker, though it should not be used alone without clinical correlation.
Microscopic Polyangiitis (MPA)
MPA lacks the granulomatous tissue destruction of GPA, presenting primarily as a pulmonary-renal syndrome. Rapidly progressive glomerulonephritis often dominates. MPO-ANCA (p-ANCA) is positive in ~60–70%. Pulmonary capillaritis causing diffuse alveolar hemorrhage is life-threatening and may require plasma exchange in addition to immunosuppression. MPA has a higher relapse rate than GPA when treated with rituximab maintenance, and cyclophosphamide-based induction is sometimes preferred for the most severe presentations.
Eosinophilic GPA (EGPA / Churg-Strauss)
EGPA unfolds in three phases: a prodromal phase with allergic rhinitis and asthma (which may precede vasculitis by years or decades), an eosinophilic phase (peripheral eosinophilia >1,500 cells/µL, eosinophilic pneumonia, eosinophilic gastroenteritis), and the vasculitic phase. Mononeuritis multiplex — sudden foot drop or wrist drop from ischemic nerve infarction — is a classic presentation. Cardiac involvement (eosinophilic myocarditis, pericarditis, cardiac tamponade) accounts for the majority of EGPA-related deaths. ANCA-positive EGPA tends to have more renal and nerve involvement; ANCA-negative EGPA has more cardiac and pulmonary eosinophilic disease. Mepolizumab (anti–IL-5) is FDA-approved for remission maintenance in EGPA.
Polyarteritis Nodosa (PAN)
PAN is distinguished from ANCA-associated vasculitis by: absence of ANCA, absence of glomerulonephritis (though renal artery involvement causes hypertension and infarction), and absence of pulmonary capillaritis. Classic presentation includes fever, weight loss, abdominal pain from mesenteric ischemia, livedo reticularis, testicular pain/tenderness, and mononeuritis multiplex. Angiography (CT or conventional) showing beaded medium-vessel aneurysms is highly suggestive. Hepatitis B testing is mandatory since HBV-associated PAN requires antiviral therapy alongside brief immunosuppression (not long-term).
IgA Vasculitis (Henoch-Schönlein Purpura)
The most common vasculitis in children, peaking at ages 4–7. Classically follows an upper respiratory infection by 1–3 weeks. The cardinal tetrad: (1) palpable purpura on the buttocks and lower extremities (pathognomonic distribution — gravity-dependent, not pressure-dependent); (2) arthritis or arthralgias of ankles and knees; (3) colicky abdominal pain, sometimes with intussusception or GI bleeding; (4) renal involvement (hematuria, proteinuria) in 20–50%, which may progress to chronic kidney disease in a small minority. Most children recover fully within 4–6 weeks; renal involvement is the main determinant of long-term prognosis. Adults with IgA vasculitis have higher rates of severe renal disease. EULAR/PReS consensus criteria require palpable purpura (mandatory) plus one of the four criteria above for diagnosis without biopsy.
Symptoms and Organ Involvement
Vasculitis can affect virtually any organ. Symptoms vary dramatically by form and vessel size, but constitutional symptoms — unexplained fever, fatigue, weight loss, and night sweats — are nearly universal in systemic vasculitis and often the first clue before organ-specific findings emerge.
Skin
Palpable purpura (non-blanching, raised red-purple spots representing leaking blood from inflamed capillaries) is the hallmark of small-vessel vasculitis. Livedo reticularis — a mottled, net-like discoloration from interrupted dermal blood flow — suggests medium-vessel disease (PAN, polyarteritis) or secondary vasculitis. Skin ulcers from digital artery occlusion, urticaria, nodules, and necrotic plaques also occur. Skin biopsy showing leukocytoclasia (neutrophil nuclear fragmentation around inflamed vessels) confirms small-vessel vasculitis histologically.
Kidneys
Glomerulonephritis is a defining feature of ANCA-associated vasculitis. Microscopic hematuria with red cell casts on urinalysis is the key finding. Proteinuria may be nephrotic range. Rising creatinine signals rapidly progressive GN — a rheumatologic emergency requiring same-day nephrology consultation and urgent biopsy. Renal biopsy showing pauci-immune crescentic GN (few immune deposits, crescentic destruction of glomeruli) is the pathological hallmark of ANCA vasculitis.
Lungs
Pulmonary capillaritis causes diffuse alveolar hemorrhage (DAH) — the most feared acute complication. Patients present with hemoptysis, falling hemoglobin, and bilateral infiltrates on imaging. Bronchoalveolar lavage returning progressively bloodier fluid confirms DAH. Pulmonary nodules in GPA can cavitate and mimic malignancy or fungal infection. Asthma is a prerequisite for EGPA. Subglottic stenosis in GPA may require laryngoscopy-guided dilation.
Nervous System
Mononeuritis multiplex — asymmetric sensorimotor deficits in the distribution of individual named peripheral nerves — results from vasculitis of the vasa nervorum feeding the nerves. Foot drop (peroneal nerve) and wrist drop (radial nerve) are classic. Cranial nerve palsies, optic neuritis, and cerebral vasculitis (headache, stroke-like episodes) occur in PAN, GCA, and CNS vasculitis. GCA-related vision loss is due to ischemia of the optic nerve or retina and is irreversible once established.
Eyes
In GCA, visual symptoms — transient monocular vision loss (amaurosis fugax), diplopia from cranial nerve palsy, or sudden permanent vision loss — constitute a medical emergency. High-dose IV methylprednisolone must be given within hours. Scleritis and uveitis occur in GPA, relapsing polychondritis, and Behçet disease. Orbital pseudotumor (proptosis from granulomatous orbital inflammation) is characteristic of GPA.
Joints and Muscles
Arthralgias and myalgias are common across all forms. Polymyalgia rheumatica (PMR) — shoulder and hip girdle aching and stiffness — coexists with GCA in 40–60% of cases. PMR without frank vasculitis is treated with lower-dose prednisone (15–20 mg vs. 40–60 mg for GCA). Synovitis resembling rheumatoid arthritis occurs in EGPA and IgA vasculitis.
Gastrointestinal Tract
Abdominal pain from mesenteric vasculitis mimics an acute abdomen. Intestinal infarction and perforation are life-threatening complications of PAN and other medium-vessel forms. GI bleeding occurs in IgA vasculitis. Intussusception in children with IgA vasculitis requires urgent surgical evaluation.
Diagnosis — Biopsy, ANCA Testing, Imaging
No single test diagnoses all forms of vasculitis. Diagnosis requires integration of clinical presentation, laboratory data, imaging, and histopathology. The urgency of pursuing tissue diagnosis is balanced against the need to start empiric treatment in life-threatening presentations (GCA threatening vision, DAH, rapidly progressive GN).
Laboratory Testing
Initial workup should include: CBC (eosinophilia in EGPA; anemia of inflammation across forms), metabolic panel and urinalysis with microscopy (creatinine, red cell casts), ESR and CRP (nonspecifically elevated; very high ESR typical of GCA), LFTs (hepatitis screen), complement levels (low C3/C4 in cryoglobulinemia, lupus vasculitis), and ANCA testing.
ANCA Testing: Two antigens and two immunofluorescence patterns are clinically important:
- PR3-ANCA (c-ANCA — cytoplasmic pattern): Highly specific for GPA (~90% sensitivity in active generalized disease). Also a disease-activity marker — rising PR3-ANCA titers predict relapse, though treatment decisions should not be based on ANCA titers alone.
- MPO-ANCA (p-ANCA — perinuclear pattern): Found in ~60–70% of MPA, ~40% of EGPA, and drug-induced vasculitis. Also elevated in many non-vasculitic conditions (IBD, endocarditis), so clinical context is essential.
Additional antibodies: ANA and anti-dsDNA (lupus), anti-GBM antibodies (Goodpasture's syndrome — distinct from but can mimic ANCA vasculitis), cryoglobulins and hepatitis serology (cryoglobulinemic vasculitis), and IgA levels (IgA vasculitis — elevated in ~50%).
Biopsy
- Temporal artery biopsy: Gold standard for GCA; biopsy ≥2 cm (skip lesions require longer segments); do not delay treatment while awaiting biopsy — steroids can be started first and biopsy remains positive for up to 2 weeks.
- Renal biopsy: Essential for characterizing glomerulonephritis (pauci-immune crescentic GN = ANCA vasculitis; IgA deposits = IgA nephropathy/IgA vasculitis; full-house deposits = lupus nephritis).
- Skin biopsy: Direct immunofluorescence showing IgA deposits in vessel walls is diagnostic of IgA vasculitis. Leukocytoclastic vasculitis on H&E confirms small-vessel inflammation.
- Lung/sinus/nerve biopsy: Granulomatous necrotizing inflammation confirms GPA. Nerve biopsy showing axonal loss with necrotizing vasculitis of epineural vessels confirms mononeuritis multiplex.
Imaging
CT Angiography and MR Angiography: Show large-vessel wall thickening (GCA, Takayasu), luminal stenosis, occlusion, or aneurysm (PAN). Aortic PET-CT can detect metabolically active large-vessel inflammation in GCA/Takayasu even when conventional angiography is normal.
High-Resolution Ultrasound: Temporal artery ultrasound showing the "halo sign" (hypoechoic thickening of the vessel wall) has comparable sensitivity to biopsy for GCA at expert centers and allows bilateral assessment. Increasingly recommended as first-line imaging.
Chest CT: Defines pulmonary nodules, cavities, ground-glass hemorrhage, and subglottic stenosis in GPA/MPA/EGPA.
Birmingham Vasculitis Activity Score (BVAS): A validated 66-item clinical scoring tool covering nine organ systems, used to quantify disease activity at diagnosis and during follow-up. BVAS scores guide treatment intensification decisions and measure remission in clinical trials. A score of 0 for ≥3 months on stable treatment defines clinical remission.
Treatment — Corticosteroids, Immunosuppressants, Biologics
Treatment goals are to induce remission rapidly, prevent organ damage, maintain remission with the lowest possible drug exposure, and manage treatment toxicity. The approach is stratified by disease severity (localized, generalized, severe/organ-threatening, immediately life-threatening) and by vasculitis type.
Remission Induction
Corticosteroids are the cornerstone of initial treatment across all forms. High-dose prednisone (1 mg/kg/day, up to 60–80 mg) is standard for GCA, ANCA vasculitis, and PAN. IV methylprednisolone (1 g/day × 3 days — "pulse steroids") is used for immediately life-threatening presentations: DAH, rapidly progressive GN with creatinine >5.7 mg/dL, threatened vision in GCA. Steroids are tapered slowly over 12–18 months; premature tapering drives relapse.
Cyclophosphamide — either IV pulse (15 mg/kg every 2–3 weeks) or daily oral — was the standard induction partner for corticosteroids in severe ANCA vasculitis for decades. It remains preferred for immediately life-threatening disease (severe DAH, creatinine >5.7 at diagnosis) and in resource-limited settings. Major toxicities: hemorrhagic cystitis, bladder cancer (cumulative dose-dependent), gonadal failure, myelosuppression, and infection. Mesna and aggressive hydration reduce bladder toxicity. Lifetime cumulative dose should be minimized.
Rituximab (anti-CD20, depletes B cells) achieved non-inferiority to cyclophosphamide for induction in the landmark RAVE trial (NEJM 2010, PMID 20461783), with superiority for relapsing GPA. Rituximab 375 mg/m² × 4 weekly doses or 1000 mg × 2 doses two weeks apart. Rituximab is preferred over cyclophosphamide in: younger patients (gonadal preservation), relapsing disease, drug-induced vasculitis, and when bladder toxicity is a concern. It is not licensed for induction in MPA in all countries — cyclophosphamide may be preferred for severe MPA with renal crisis.
Plasma Exchange (PLEX/Plasmapheresis): Previously used widely for severe ANCA vasculitis with advanced renal failure, the PEXIVAS trial (NEJM 2020) showed plasma exchange did not reduce the primary composite outcome of death or kidney failure at 7 years vs. immunosuppression alone, overturning prior practice. PLEX remains an option in anti-GBM co-positivity and in DAH refractory to pulse steroids, but is no longer routine for ANCA vasculitis with severe renal disease.
Remission Maintenance
After induction (typically 3–6 months), maintenance therapy switches to lower-toxicity regimens:
- Azathioprine (2 mg/kg/day) — first-line maintenance for MPA and GPA after cyclophosphamide induction; check TPMT enzyme activity before starting.
- Methotrexate (20–25 mg/week) — alternative for non-renal GPA; not appropriate when GFR <60.
- Rituximab maintenance — 500 mg every 6 months for 24 months is now preferred over azathioprine for GPA and MPA based on MAINRITSAN trials. Reduces relapse rates from ~30% to ~5% at 28 months.
- Mycophenolate mofetil (MMF) — an alternative for maintenance when azathioprine and methotrexate are not tolerated; less evidence than the above agents.
- Avacopan (CCX168) — a selective complement C5a receptor inhibitor approved in 2021 for ANCA vasculitis. In the ADVOCATE trial, it achieved comparable remission induction to high-dose prednisone when combined with rituximab or cyclophosphamide, with significantly less steroid exposure and better kidney function recovery at 52 weeks. An important advance for steroid-sparing induction.
Biologics for Giant Cell Arteritis
Tocilizumab (anti-IL-6 receptor, Actemra) is FDA-approved for GCA based on the GiACTA trial (NEJM 2017, PMID 28745999). Weekly subcutaneous 162 mg achieved sustained remission in 56% of patients at 52 weeks vs. 18% with 26-week prednisone taper and 14% with 52-week taper. Prednisone could be tapered to zero in most tocilizumab-treated patients, dramatically reducing steroid toxicity. Monitor for neutropenia, elevated LFTs, and bowel perforation (rare but serious). Biological monitoring of GCA activity is complicated because tocilizumab suppresses IL-6 and hence normalizes ESR/CRP regardless of disease activity — imaging (PET-CT, ultrasound) becomes important for assessing large-vessel activity on tocilizumab.
Mepolizumab (anti-IL-5) is approved for remission maintenance in EGPA, reducing the relapse rate and allowing steroid tapering in patients with relapsing or refractory disease.
Treatment of Secondary Vasculitis
HBV-associated PAN: antiviral therapy (entecavir or tenofovir) combined with brief plasma exchange and short-course steroids — prolonged immunosuppression promotes viral replication. HCV-associated cryoglobulinemic vasculitis: direct-acting antivirals (DAAs) achieve viral clearance in >95% and resolve vasculitis in most patients; rituximab for severe acute manifestations. Drug-induced vasculitis: stop the offending drug; short-course steroids for severe cases.
Natural and Nutritional Approaches
Natural and nutritional strategies cannot replace immunosuppressive therapy for active vasculitis but can meaningfully support conventional treatment, reduce cardiovascular risk (which is substantially elevated in vasculitis), mitigate steroid toxicity, and maintain remission in patients on stable maintenance therapy. Always discuss supplements with your rheumatologist, as some immune-modulating supplements can theoretically interact with immunosuppressants or influence disease activity monitoring.
Omega-3 Fatty Acids
Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) from oily fish (salmon, sardines, mackerel) or fish oil supplements reduce pro-inflammatory leukotriene B4 (LTB4) and thromboxane A2 production, both of which drive neutrophil-mediated vascular inflammation. Several small studies in GPA showed modest reductions in relapse rates with high-dose fish oil (3–4 g EPA+DHA/day). In EGPA, omega-3s may help reduce eosinophil-driven inflammation. Aim for 2–3 servings of oily fish per week or discuss supplementation with your doctor.
Anti-Inflammatory Diet
A Mediterranean-style diet — rich in vegetables, olive oil, legumes, nuts, whole grains, and oily fish — reduces circulating IL-6, TNF-alpha, and CRP. These are the same cytokines that drive GCA and ANCA vasculitis. While no randomized controlled trial of dietary intervention in vasculitis exists, the biological plausibility is strong and the cardiovascular benefit is well-established. Patients on long-term corticosteroids especially benefit from reducing refined carbohydrate and saturated fat intake to counter steroid-induced dyslipidemia and insulin resistance.
Vitamin D
Vitamin D deficiency is common in autoimmune vasculitis and associated with more severe disease and higher relapse rates in some cohort studies. Vitamin D has immunomodulatory effects — promoting regulatory T cells and reducing Th17 activity, the same pathway implicated in GCA. Patients on long-term corticosteroids require vitamin D supplementation (1,000–2,000 IU/day minimum, with calcium) to prevent steroid-induced osteoporosis. Target serum 25-OH vitamin D of 40–60 ng/mL (100–150 nmol/L).
Smoking Cessation
Smoking is associated with increased severity of EGPA, higher ANCA titers in GPA, and substantially elevated cardiovascular risk across all vasculitis forms. Patients with GCA and large-vessel involvement have a 3-fold increased risk of aortic aneurysm — smoking compounds this risk. Cessation is the single most impactful lifestyle modification for vasculitis patients who smoke.
Antioxidant Foods and Supplements
Oxidative stress amplifies vascular inflammation. A diet rich in antioxidants — colorful vegetables and fruits (berries, leafy greens, cruciferous vegetables), green tea, and turmeric (curcumin) — may reduce inflammatory burden. Vitamin C (500–1000 mg/day) and vitamin E (400 IU/day) have been studied in inflammatory vascular conditions with modest effects on endothelial function. Quercetin and resveratrol have anti-inflammatory properties in cell studies, though human trial data in vasculitis are limited.
Bone and Muscle Health on Steroids
Long-term corticosteroid use causes osteoporosis, avascular necrosis, and muscle weakness (steroid myopathy). Weight-bearing exercise, calcium (1,200 mg/day from food and supplements combined), vitamin D, and bisphosphonates (alendronate, risedronate) are standard preventive measures for patients on prednisone >5 mg/day for >3 months. FRAX score calculation for fracture risk is recommended at the start of long-term steroid therapy.
Stress Reduction
Psychological stress activates the HPA axis and can promote inflammatory cytokine release. Mindfulness meditation, gentle yoga, and structured relaxation techniques may help reduce flare frequency in autoimmune conditions, though evidence specific to vasculitis is anecdotal. Sleep hygiene is particularly important — corticosteroids disrupt sleep architecture, and poor sleep amplifies inflammatory signaling.
Disease Course and Prognosis
Vasculitis follows highly variable disease courses depending on the form, organ involvement at presentation, rapidity of diagnosis, and response to treatment. Modern immunosuppressive regimens have transformed previously fatal conditions into manageable chronic diseases for most patients.
ANCA-Associated Vasculitis
Five-year survival has improved from <20% in the pre-cyclophosphamide era to >75–80% with modern treatment. The leading causes of death are now treatment-related infections (especially in the first year), cardiovascular events, and end-stage renal disease rather than uncontrolled vasculitis itself. Relapse remains the central challenge — GPA relapses in 50–70% of patients within 5 years; MPA relapses in 30–40%. PR3-ANCA positivity, GPA (vs. MPA), nasal carriage of S. aureus, and residual renal impairment at remission predict relapse. Long-term rituximab maintenance dramatically reduces relapse rates.
Giant Cell Arteritis
GCA does not shorten life expectancy when managed appropriately, but permanent vision loss occurs in 15–20% of patients who delay treatment. Large-vessel involvement (aortic aneurysm, limb ischemia) is a late-emerging risk that requires lifelong vascular surveillance with imaging every 1–2 years even in remission. Most patients require steroids for 2–4 years; some require indefinite low-dose maintenance. Tocilizumab-enabled steroid tapering has substantially reduced the burden of steroid toxicity (osteoporosis, diabetes, cataracts, cardiovascular events).
IgA Vasculitis
In children, >95% recover fully within 4–6 weeks. Renal involvement requiring long-term follow-up occurs in 1–5%. Adults have higher rates of persistent renal disease and a smaller proportion achieve complete resolution. Post-strep cases rarely recur after the inciting infection resolves.
PAN and Takayasu Arteritis
PAN, when not associated with hepatitis B, follows a relapsing-remitting course managed with steroids and sometimes cyclophosphamide. HBV-PAN, treated with antivirals, often achieves durable remission without ongoing immunosuppression. Takayasu arteritis in young women can cause decades of vascular complications including limb ischemia, hypertension, and stroke; management involves steroids, steroid-sparing agents (methotrexate, azathioprine, tocilizumab), and vascular reconstruction procedures for critical stenoses.
Complications and Organ Damage
Vasculitis causes organ damage through two distinct mechanisms: active inflammation destroying vessel walls and occluding blood flow, and treatment toxicity from prolonged immunosuppression. The Vasculitis Damage Index (VDI) is a validated tool tracking cumulative irreversible damage across 11 organ systems.
Renal Failure
End-stage renal disease (ESRD) requiring dialysis or transplantation affects 20–25% of patients with ANCA-associated vasculitis within 5 years. Creatinine at presentation, the degree of fibrous crescent formation on biopsy (irreversible fibrosis vs. active cellular crescents), and delay to treatment are the strongest predictors. Patients with ANCA vasculitis who reach ESRD can receive kidney transplants after adequate time in remission — vasculitis recurrence in the transplant is rare (<5%) and manageable.
Pulmonary Damage
Pulmonary fibrosis following repeated episodes of alveolar hemorrhage or eosinophilic pneumonia causes irreversible restrictive lung disease. Subglottic stenosis in GPA may require repeated bronchoscopic dilation or tracheostomy. Pulmonary hypertension can develop as a late complication.
Cardiovascular Events
Systemic vasculitis increases the risk of myocardial infarction and stroke 2- to 3-fold above the general population — a combination of disease-driven endothelial dysfunction, accelerated atherosclerosis, and steroid-induced dyslipidemia and hypertension. Aortic aneurysm — particularly in GCA and Takayasu — can rupture fatally. Annual blood pressure, lipid, and glucose monitoring with aggressive cardiovascular risk factor management is mandatory.
Peripheral Neuropathy
Mononeuritis multiplex, if not treated promptly, results in irreversible axonal loss and permanent foot drop, wrist drop, or sensory loss. Physical therapy and orthotic devices help with rehabilitation, but the nerve damage itself does not reverse once established.
Vision Loss
Permanent visual loss in GCA, once established, does not recover with steroids. Bilateral involvement occurs in up to 20% of cases with unilateral initial loss if treatment is not started immediately. Vision loss in the other eye is prevented — not reversed — by high-dose steroids.
Treatment Toxicity
Corticosteroids cause: osteoporosis (20–40% of long-term users develop fractures), cataracts, glaucoma, diabetes, hypertension, adrenal suppression, Cushing's syndrome, and avascular necrosis of the hip. Cyclophosphamide: bladder cancer (3–5% cumulative risk with prolonged use), premature ovarian failure or azoospermia, myelodysplastic syndrome. Methotrexate: hepatotoxicity, pulmonary hypersensitivity. Rituximab: hypogammaglobulinemia with recurrent infections, rare progressive multifocal leukoencephalopathy (PML), and prolonged B-cell depletion requiring immunoglobulin replacement in some patients.
Prevention and Monitoring
While primary prevention of vasculitis is not possible given its largely idiopathic nature, secondary prevention of organ damage, relapse, and treatment toxicity is achievable with structured monitoring.
Infection Prophylaxis During Immunosuppression
Patients on cyclophosphamide or high-dose steroids require trimethoprim-sulfamethoxazole (TMP-SMX) prophylaxis against Pneumocystis jirovecii pneumonia (PCP). Patients on rituximab should be screened for hepatitis B (HBsAg, anti-HBc) before starting — rituximab can reactivate occult hepatitis B, requiring prophylactic antiviral therapy. Annual influenza vaccination and pneumococcal vaccination are recommended; live vaccines (MMR, varicella, yellow fever) are contraindicated during significant immunosuppression. Monitor IgG levels on rituximab — levels <4 g/L indicate need for IVIG replacement therapy.
Disease Activity Monitoring
Urinalysis with microscopy should be performed at every visit for patients with ANCA vasculitis — microscopic hematuria returning during remission is the earliest sign of renal relapse. Monthly ANCA titers during the first year, with rising titers prompting close clinical surveillance (though not automatically triggering preemptive treatment escalation, per current guidelines). GCA patients on tocilizumab require imaging-based activity assessment (PET-CT or MRA) since inflammatory markers are suppressed by the drug regardless of disease activity.
Cardiovascular Risk Reduction
Blood pressure target <130/80 mmHg. LDL-cholesterol target <70 mg/dL in patients with established cardiovascular risk factors. Low-dose aspirin for patients with prior cardiovascular events. Glucose monitoring for steroid-induced hyperglycemia — many patients require short-term or long-term antidiabetic therapy during steroid courses.
Bone Protection
FRAX-guided assessment at start of any steroid course expected to last >3 months. Calcium 1,200 mg/day + vitamin D 1,000–2,000 IU/day from initiation of steroids. Bisphosphonate (alendronate 70 mg/week or risedronate 35 mg/week) for patients at high fracture risk or on steroids long-term. DEXA scan at baseline and every 1–2 years.
Relapse Recognition and Patient Education
Patients should be educated to report immediately: return of sinusitis or nasal symptoms (GPA), hemoptysis or new cough, new hematuria noted on urine dipstick, visual symptoms (GCA), or constitutional symptoms (fever, night sweats, weight loss). Home urine dipstick testing kits allow early self-detection of hematuria between clinic visits. Patient support organizations such as the Vasculitis Foundation provide peer support, self-management resources, and disease-specific education materials.
Key Research Papers
- Jennette JC et al., 2013 — 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides — PMID: 23045170
- Search PubMed
- Stone JH et al., 2017 — Tocilizumab in Giant Cell Arteritis (GiACTA Trial), NEJM — PMID: 28745999
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Zwerina J et al., 2009 — Eosinophilic Granulomatosis with Polyangiitis (Churg-Strauss Syndrome): Overview of Clinical Manifestations and Treatment — PMID: 18782791
- Search PubMed
- Search PubMed
Connections
- Rheumatology
- Rheumatoid Arthritis
- Arthritis
- Lupus
- Raynaud's Disease
- Sjögren's Syndrome
- Ankylosing Spondylitis
- Gout
- Osteoarthritis
- Ehlers-Danlos Syndrome
- Osteoporosis
- Cardiovascular Disease
- ANCA Test — the antibody test that confirms ANCA-associated vasculitis and separates PR3 from MPO disease.
- Microscopic Polyangiitis (MPA) — ANCA-associated small-vessel vasculitis defined by the absence of granulomas.
- Granulomatosis with Polyangiitis (GPA) — ANCA-associated vasculitis with necrotizing granulomas of the airways and kidneys.
- Glomerulonephritis — the kidney manifestation of ANCA-associated and immune-complex vasculitis.
- Henoch-Schönlein Purpura (IgA Vasculitis) — the most common vasculitis of childhood, covered in full on its own page.
- Granulomatosis with Polyangiitis (Rheumatology) — the rheumatology view of the same ANCA-associated vasculitis, with induction and maintenance regimens.