Henoch-Schönlein Purpura (IgA Vasculitis)
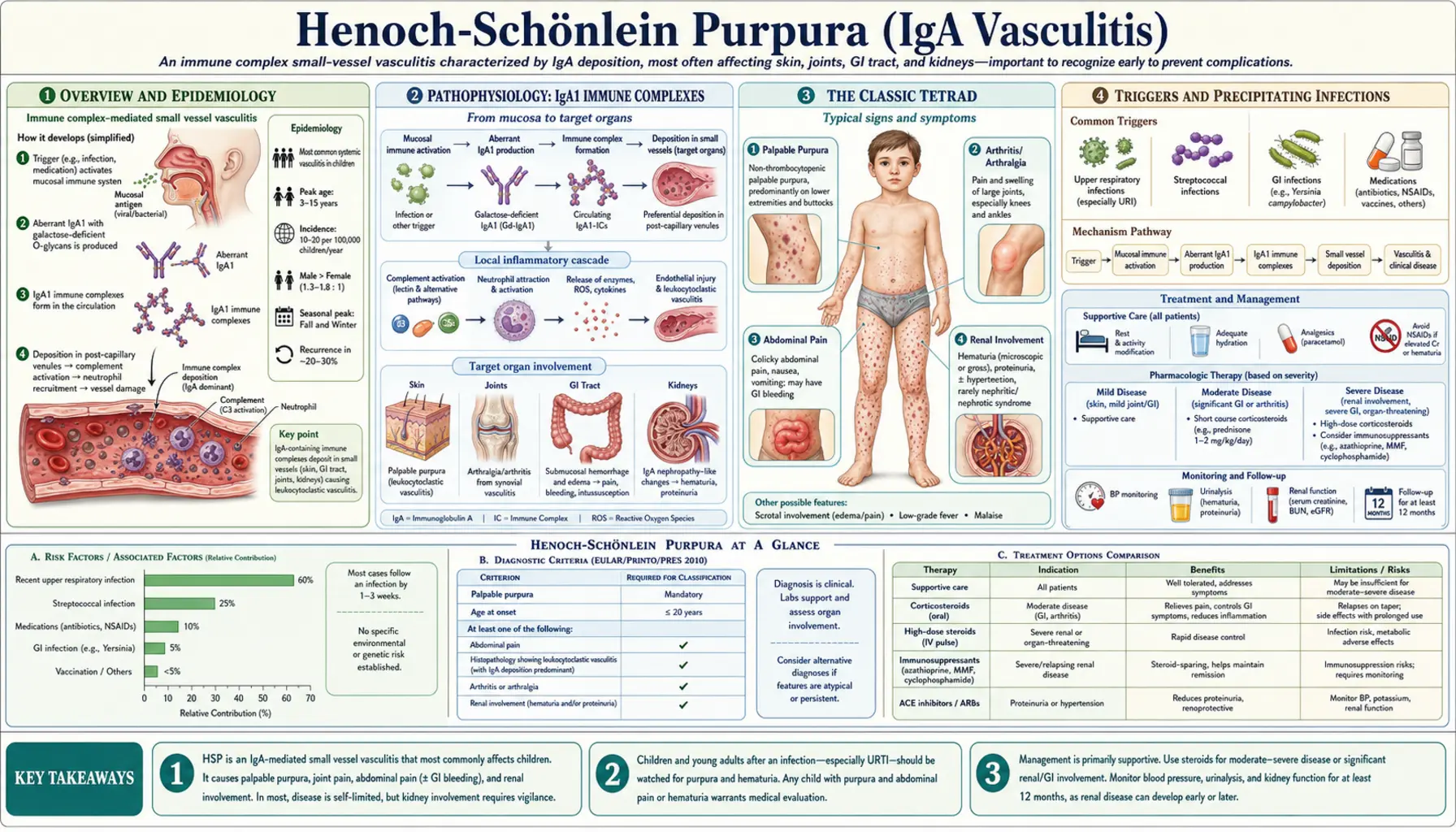
Henoch-Schönlein Purpura (HSP), now formally called IgA Vasculitis (IgAV), is the most common systemic vasculitis of childhood. It is driven by IgA1-dominant immune complex deposition in the walls of small blood vessels throughout the body — producing the classic tetrad of palpable purpura on the lower extremities, arthritis, colicky abdominal pain, and kidney inflammation. Although it sounds alarming, the vast majority of children recover fully within weeks. The critical job is recognizing it promptly, managing pain and joint swelling, and monitoring the kidneys carefully for the minority who develop lasting renal disease.
Table of Contents
- Overview and Epidemiology
- Pathophysiology: IgA1 Immune Complexes
- The Classic Tetrad
- Triggers and Precipitating Infections
- Diagnosis: Clinical and Laboratory
- Renal Involvement and Prognosis
- Treatment and Management
- HSP in Adults vs. Children
- Key Research Papers
- Connections
- Featured Videos
Overview and Epidemiology
IgA Vasculitis affects approximately 10–20 per 100,000 children per year in the developed world, with a peak incidence between ages 3 and 15. Boys are affected slightly more often than girls (roughly 1.5:1). The condition is distinctly seasonal — case clusters peak in autumn and winter, mirroring the surge in upper respiratory infections that typically precede it by 1–3 weeks.
The disease was described by Johann Schönlein in 1837, who linked purpura with joint involvement, and later expanded by Eduard Henoch in 1874, who added the gastrointestinal and renal components. In 2012, the European League Against Rheumatism (EULAR) and Paediatric Rheumatology European Society (PRES) renamed it IgA Vasculitis to reflect its molecular mechanism rather than its eponymous history — but most clinicians and patients still call it HSP.
The good news: roughly 94% of affected children achieve complete remission. Recurrences occur in approximately 30–40% of cases, but these are generally milder than the initial episode and resolve without escalating treatment.
Pathophysiology: IgA1 Immune Complexes
The central defect in IgA Vasculitis is the overproduction of aberrantly glycosylated IgA1. Healthy IgA1 molecules carry sugar chains (O-glycans) that include galactose on their hinge region. In HSP patients, many of these IgA1 molecules are produced with a galactose-deficient hinge — they are called "Gd-IgA1." This structural abnormality makes Gd-IgA1 molecules sticky: they aggregate into immune complexes and are poorly cleared by the liver.
These circulating Gd-IgA1 immune complexes deposit in the walls of small blood vessels — including those of the skin, joints, gut, and kidney glomeruli. Once deposited, they activate complement (via the alternative and lectin pathways) and recruit neutrophils and macrophages, producing the leukocytoclastic vasculitis (vessel inflammation with white blood cell infiltration) that underlies every symptom of the disease.
In the kidney, IgA1 deposits accumulate in the mesangium — the support tissue of the glomerular tuft — causing the identical pathological lesion seen in adult IgA nephropathy (Berger's disease). This molecular connection explains why HSP renal disease and IgA nephropathy are considered spectrum diseases sharing the same IgA1 glycosylation defect, just presenting at different ages and with different extents of systemic involvement.
The Classic Tetrad
The diagnosis of HSP / IgA Vasculitis is clinical. Four hallmark features constitute the classic tetrad:
1. Palpable Purpura
The defining feature: crops of raised, non-blanching, red-to-purple spots — true palpable purpura — distributed predominantly on the lower extremities and buttocks. The gravity-dependent pattern is characteristic: gravity concentrates hydrostatic pressure in the dependent small vessels, driving immune complex deposition there preferentially. The extensor surfaces of the arms and the periarticular areas around the knees and ankles are also commonly involved. The face and trunk are usually spared. Crucially, the purpura are non-thrombocytopenic: the platelet count is normal or elevated. This distinguishes HSP from idiopathic thrombocytopenic purpura (ITP) and meningococcemia.
Lesions evolve through stages — beginning as urticarial wheals or erythematous macules, progressing to raised purpura, then browning as they age. New crops often appear over days to weeks, especially if the child is on their feet. Bed rest visibly reduces new lesion formation by reducing hydrostatic pressure.
2. Arthritis or Arthralgia
Joint involvement occurs in 60–80% of children with HSP. The large joints — knees and ankles — are predominantly affected in a migratory, oligoarticular pattern. Joint swelling and pain can be severe enough to limit walking, but it is periarticular rather than true synovitis in most cases, meaning the joint space is not destructively involved. Permanent joint damage does not occur. Arthritis often precedes the rash by 1–2 days in some patients, which can initially lead to diagnostic confusion.
3. Abdominal Pain
Gastrointestinal involvement occurs in 50–75% of children. Colicky, periumbilical or diffuse abdominal pain results from IgA-immune-complex vasculitis of the gut wall, causing submucosal edema and hemorrhage. Children may have nausea, vomiting, and bloody stools. GI bleeding is typically occult (positive stool guaiac), though overt rectal bleeding can occur. The most feared GI complication is intussusception — HSP-associated intussusception occurs at the small bowel (ileoileal), unlike the more common ileocolic intussusception in idiopathic cases, and is a lead-point intussusception driven by submucosal edema. Ultrasound is the preferred diagnostic tool; air or saline enema reduction may fail for small-bowel intussusception, requiring surgical intervention in some cases.
4. Renal Involvement
Kidney disease occurs in 40–50% of children, typically presenting 4–6 weeks after the rash onset — sometimes after the skin and joint disease has resolved. Manifestations range from microscopic hematuria (the most common, often transient) to frank nephritic or nephrotic syndrome. Renal biopsy shows mesangial IgA deposits on immunofluorescence — the hallmark of IgA nephropathy. Prognosis is generally favorable for children with isolated hematuria, but those with significant proteinuria, nephrotic range losses, or severe hypertension are at risk for progressive renal disease. Approximately 1–2% of children with HSP develop end-stage renal disease over long-term follow-up.
Triggers and Precipitating Infections
An identifiable upper respiratory tract infection precedes HSP by 1–3 weeks in roughly two-thirds of cases. The most commonly implicated pathogens include:
- Group A Streptococcus (GAS): The best-documented trigger; streptococcal antigens can act as molecular mimics or direct immune stimulants that upregulate aberrant IgA1 glycosylation.
- Adenovirus and other respiratory viruses: Frequent temporal associations, though direct causality is harder to establish.
- Parvovirus B19: Associated with several pediatric case series, particularly in younger children.
- Helicobacter pylori: Implicated in some adult cases and in recurrent pediatric HSP, though evidence is less consistent.
Vaccines, drugs (penicillin, ACE inhibitors, NSAIDs), and insect bites have been reported as triggers in individual cases. COVID-19 infection has been associated with HSP-like presentations, though the relationship to MIS-C (multisystem inflammatory syndrome in children) remains under investigation — MIS-C shares some clinical overlap but involves different immune pathways (IgG-dominant vs. IgA-dominant).
Diagnosis: Clinical and Laboratory
There is no single confirmatory laboratory test for HSP. The 2010 EULAR/PRES/PRINTO classification criteria require palpable purpura (mandatory) plus at least one of four supporting criteria:
- Diffuse abdominal pain (colicky, acute onset)
- Arthritis (acute; any joint) or arthralgia without joint destruction
- Renal involvement (proteinuria, hematuria, red blood cell casts)
- Leukocytoclastic vasculitis or proliferative glomerulonephritis with predominant IgA deposition on biopsy
In a child with the classic tetrad, the diagnosis is made clinically and biopsy is unnecessary. Skin biopsy — when performed — shows IgA immunofluorescence staining of small vessel walls, confirming the diagnosis.
Typical laboratory findings:
- Normal platelet count (distinguishes from ITP and meningococcemia)
- Mildly elevated WBC, CRP, and ESR — nonspecific inflammation markers
- Elevated serum IgA in 50–70% of patients (not required for diagnosis)
- Urinalysis: hematuria ± proteinuria (check at presentation and weekly for 6 weeks)
- Normal complement levels (C3, C4) — helps distinguish from lupus vasculitis
- Anti-streptolysin O (ASO) titer if GAS trigger is suspected
Imaging: Abdominal ultrasound if GI involvement is significant — to evaluate for intussusception, bowel wall thickening, and free fluid. Scrotal ultrasound if testicular pain is present (orchitis occurs in ~10–15% of boys with HSP; must be differentiated from testicular torsion).
Renal Involvement and Prognosis
Renal involvement is the principal determinant of long-term outcome in HSP. It is essential to monitor urine for hematuria and proteinuria at every visit during the acute phase and for at least 6 months after the rash resolves, because nephritis can appear weeks after all other symptoms have cleared.
Risk stratification:
- Isolated microscopic hematuria: excellent prognosis; >95% resolve without intervention
- Hematuria + mild proteinuria: generally resolves; monitor closely
- Nephritic syndrome (hematuria + hypertension + RBC casts + rising creatinine): requires nephrology referral and renal biopsy
- Nephrotic-range proteinuria (>3.5 g/day in adults; >40 mg/m²/hr in children): higher risk of progressive CKD
- Combined nephritic-nephrotic: worst renal prognosis; 10–20% progress to CKD or ESRD over decades
Renal biopsy indications include: nephrotic-range proteinuria, rapidly rising creatinine, hypertension requiring treatment, or any suspected severe nephritis. Histology is classified by the ISKDC (International Study of Kidney Disease in Children) grading system: Grade I (minimal changes) through Grade VI (crescentic involvement >75%). High-grade lesions (IV–VI) receive aggressive immunosuppression.
Long-term renal monitoring is important even in apparently recovered patients. Studies following HSP children for 10–23 years show that 1–2% develop ESRD and approximately 20% of those with initial significant renal involvement have hypertension or reduced GFR at long-term follow-up. Pregnancy can unmask latent HSP nephropathy in women who appeared to recover fully as children.
Treatment and Management
Mild disease (purpura + arthralgia only):
- Supportive care: hydration, rest, elevation of affected limbs
- NSAIDs (ibuprofen) for joint pain and fever — use with caution if renal involvement is suspected, as NSAIDs reduce glomerular filtration
- Acetaminophen is a safer analgesic if renal involvement is present or uncertain
Moderate-to-severe abdominal pain or arthritis:
- Short-course oral corticosteroids (prednisone 1 mg/kg/day for 1–2 weeks, then taper) significantly reduce the severity and duration of abdominal pain and joint involvement
- Evidence is mixed on whether steroids prevent renal involvement — most meta-analyses show no benefit for renal protection in unselected patients, but early steroids for severe GI disease are widely practiced
Significant renal disease:
- ACE inhibitors or ARBs: first-line for proteinuria reduction and renoprotection
- High-dose pulse IV methylprednisolone followed by oral prednisone for rapidly progressive nephritis
- Combination immunosuppression (azathioprine, mycophenolate mofetil, or cyclophosphamide) for ISKDC Grade IV–VI or crescentic nephritis, guided by nephrology
- Rituximab: emerging evidence for refractory cases
Recurrent HSP: Up to 40% of children have at least one recurrence. Recurrences are usually milder and managed with the same supportive approach. Identifying and treating an underlying streptococcal carrier state may reduce recurrence frequency.
HSP in Adults vs. Children
IgA Vasculitis in adults (roughly 10% of all HSP cases) follows a substantially more severe course than in children:
- Renal involvement is more frequent and more severe: up to 75% of adults develop nephritis vs. 40–50% of children
- Up to 30–40% of adults with HSP nephritis progress to CKD over long-term follow-up
- Gastrointestinal complications including hemorrhage and bowel necrosis are more common
- The purpuric rash is often more extensive and may involve the trunk and upper extremities
- Adults are more likely to require immunosuppressive therapy beyond steroids
The reason for the age-dependent severity difference is incompletely understood but may relate to differences in IgA1 glycosylation capacity, pre-existing subclinical renal damage, or differences in complement activation efficiency between mature and developing immune systems.
Key Research Papers
- Search PubMed — Established the first consensus classification criteria for childhood vasculitides, including the original criteria for HSP that distinguish it from other purpuric conditions.
- Search PubMed — Updated 2010 classification criteria (the currently used standard); validated in a multicenter international cohort; palpable purpura as mandatory criterion with one additional feature required.
- Search PubMed — International consensus renaming HSP to "IgA Vasculitis (Henoch-Schönlein)" and formally linking it to IgA nephropathy as a spectrum disease.
- Search PubMed — Systematic review finding that while corticosteroids reduce abdominal pain duration and severity, evidence does not support their use to prevent nephritis in unselected HSP patients.
- Search PubMed — Comprehensive review of the IgA1 galactose-deficiency mechanism, Gd-IgA1 immune complex formation, complement activation pathways, and how these drive multiorgan involvement.
- Search PubMed — Practical pediatric review covering clinical presentation, diagnosis, management, and monitoring protocols; widely cited for its straightforward evidence-based management summary.
- Search PubMed — Early landmark study defining the spectrum of renal manifestations in HSP and establishing that the severity of nephritis, not the rash or joint disease, determines long-term prognosis.
- Search PubMed — Meta-analysis of 12 studies (1133 children) demonstrating that children with only microscopic hematuria have <1% risk of long-term renal damage, supporting a risk-stratified follow-up strategy.
- Search PubMed — Landmark Finnish follow-up study of 141 HSP children over 23 years: 1.8% developed ESRD, 79% had complete resolution; identified nephrotic-range proteinuria as the strongest predictor of poor renal outcome.
- Search PubMed — Retrospective cohort of 150 Italian children detailing frequency of each component of the tetrad, recurrence rates (~33%), and outcomes, with emphasis on the importance of serial urinalysis monitoring.
- Search PubMed — Largest adult HSP cohort study; confirms substantially more severe renal involvement in adults (75% nephritis) and worse long-term renal outcomes compared to pediatric disease.
- Search PubMed — Contemporary review highlighting unresolved questions in HSP management including corticosteroid use, the role of renin-angiotensin blockade for early nephropathy, and emerging insights from genomics of IgA1 glycosylation defects.
Connections
- Pediatrics
- Kawasaki Disease
- Intussusception
- IgA Nephropathy
- Rheumatology
- Nephrology
- Lupus (SLE)
- Febrile Seizures
- Vasculitis — the wider family of blood-vessel inflammation that HSP belongs to, and how the other forms differ.