Traumatic Brain Injury (TBI)
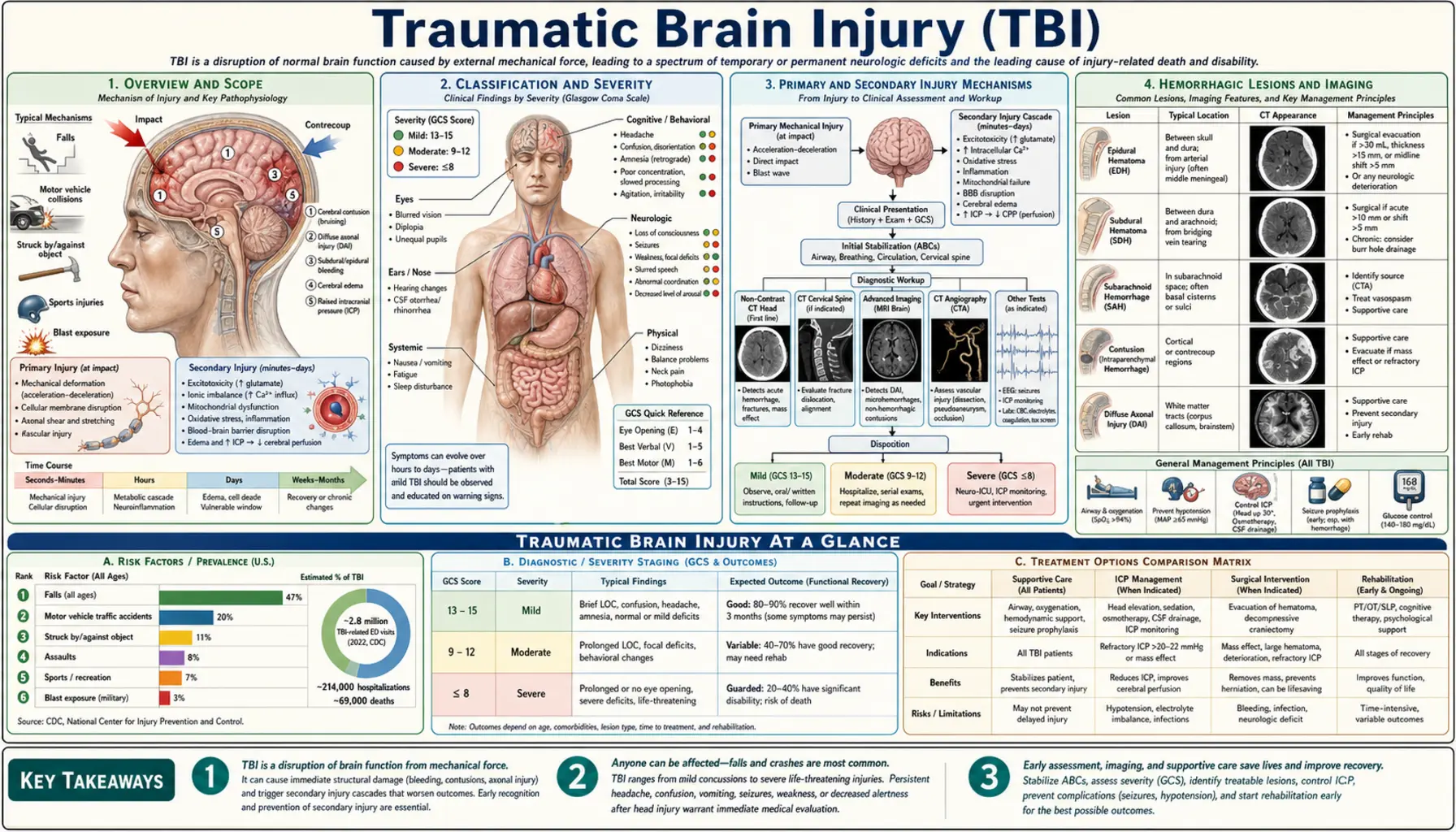
Table of Contents
- Overview and Scope
- Classification and Severity
- Primary and Secondary Injury Mechanisms
- Hemorrhagic Lesions and Imaging
- Acute Emergency Management
- Intracranial Pressure Monitoring and Control
- Neurosurgical Interventions
- ICU Care and Systemic Management
- Rehabilitation and Long-Term Outcomes
- Prevention
- Research Papers
- Connections
- Featured Videos
Overview and Scope
Traumatic brain injury (TBI) is an alteration in brain function — or other evidence of brain pathology — caused by an external mechanical force. The term spans a wide continuum from a brief concussion that resolves in days to a catastrophic injury that leaves a person in a vegetative state. This page focuses on moderate-to-severe TBI (Glasgow Coma Scale ≤12), where structural brain damage, hemorrhage, cerebral edema, and elevated intracranial pressure are central concerns. For mild TBI (concussion), see the Concussion page.
TBI is one of the leading causes of death and disability worldwide. In the United States, approximately 1.5 million Americans sustain a TBI each year, resulting in roughly 230,000 hospitalizations and 50,000 deaths. Falls account for nearly half of all TBI-related emergency department visits, followed by motor vehicle accidents, being struck by or against an object, and assault. Blast injuries from explosive devices are the signature TBI mechanism in military conflicts and present with unique pathophysiology distinct from blunt civilian trauma.
Despite advances in neurocritical care, moderate-to-severe TBI remains a condition with limited direct pharmacological treatment. Outcomes are heavily determined by the speed and precision of physiological stabilization in the first hours after injury — preventing the secondary brain injury cascade is the central task of acute management.
Classification and Severity
TBI severity is classified primarily by the Glasgow Coma Scale (GCS), a structured neurological assessment measuring eye opening, verbal response, and motor response. Scores range from 3 (no response) to 15 (fully alert).
Severity Categories
- Mild TBI (concussion): GCS 13–15; no loss of consciousness or loss lasting <30 minutes; post-traumatic amnesia <24 hours; normal CT scan in most cases. See Concussion.
- Moderate TBI: GCS 9–12; loss of consciousness <6 hours; post-traumatic amnesia 1–7 days; CT may show structural abnormalities. These patients require hospitalization and close monitoring for neurological deterioration.
- Severe TBI: GCS ≤8; loss of consciousness ≥6 hours or coma; post-traumatic amnesia >7 days; structural injury on CT is common. Patients typically require intensive care, often intubation, and may need neurosurgical intervention.
- Complicated mild TBI: GCS 13–15 (mild category) but with abnormal findings on CT — a clinically important subgroup that behaves more like moderate TBI.
Structural Classification
- Focal injury: damage concentrated in one area — contusions, hematomas, penetrating wounds. More likely in moderate-severe TBI from blunt impact.
- Diffuse injury: widespread axonal stretching and disconnection across brain regions — diffuse axonal injury (DAI). The hallmark of acceleration-deceleration mechanisms; often severe despite a relatively unremarkable early CT.
- Mixed focal and diffuse: the most common pattern in severe TBI.
Marshall CT Classification
The Marshall Classification categorizes TBI on CT into six categories (Diffuse Injury I through IV, Evacuated Mass Lesion, Non-Evacuated Mass Lesion) based on whether cisterns are compressed, midline is shifted, and whether a high-density lesion is present and evacuated. Higher Marshall scores correlate with worse outcomes and guide urgency of neurosurgical consultation.
Primary and Secondary Injury Mechanisms
TBI pathophysiology unfolds in two overlapping phases that are conceptually distinct but clinically inseparable.
Primary Injury
Primary injury is the mechanical damage that occurs at the moment of impact. It is immediate and largely irreversible. The forces involved — contact forces (compression, laceration, skull fracture) and inertial forces (rapid acceleration-deceleration, rotation) — produce:
- Cortical contusions (bruises on the brain surface, classically at frontal and temporal poles)
- Diffuse axonal injury (stretching and shearing of white-matter tracts, particularly corpus callosum, dorsolateral brainstem, and periventricular regions)
- Vascular disruption (tearing of bridging veins, meningeal arteries, or parenchymal vessels producing hemorrhage)
- Neuronal and glial death at the impact site
Nothing can undo primary injury. However, the hours and days after injury offer a critical window to limit secondary injury.
Secondary Injury
Secondary injury refers to the cascade of biochemical, cellular, and systemic events triggered by primary injury that progressively expand the zone of damage. Key mechanisms include:
- Cerebral edema: both cytotoxic edema (cell swelling from ion pump failure) and vasogenic edema (blood-brain barrier breakdown) increase brain volume, raising intracranial pressure (ICP). Severe edema with ICP >20–25 mmHg impairs cerebral perfusion.
- Excitotoxicity: massive glutamate release overactivates NMDA and AMPA receptors, flooding neurons with calcium ions that trigger apoptotic and necrotic cascades.
- Oxidative stress: reactive oxygen species from mitochondrial dysfunction and free-hemoglobin metabolism damage lipid membranes, proteins, and DNA.
- Neuroinflammation: activated microglia and infiltrating peripheral immune cells release cytokines (IL-1β, TNF-α, IL-6) that amplify tissue damage over days to weeks.
- Ischemia: reduced cerebral blood flow from elevated ICP, cerebral vasospasm, and systemic hypotension combines with the metabolic demands of neuroinflammation to create regions of secondary ischemia around the primary injury core.
- Mitochondrial dysfunction: impaired oxidative phosphorylation reduces ATP production precisely when cellular energy demand is highest, accelerating cell death.
The secondary injury cascade is the main target of neurocritical care. Every intervention in the ICU — maintaining blood pressure, avoiding hypoxia, controlling ICP, preventing fever — is aimed at interrupting one or more of these pathways.
Hemorrhagic Lesions and Imaging
Identifying and characterizing intracranial hemorrhage is one of the first priorities after moderate-to-severe TBI. CT without contrast is the standard initial imaging modality because it is fast, widely available, and highly sensitive for acute blood.
Epidural Hematoma (EDH)
An epidural hematoma is a collection of blood between the skull and the dura mater. On CT it appears as a biconvex (lens-shaped) hyperdense (white) mass that does not cross suture lines because the dura is tightly adherent at sutures. It is most commonly caused by rupture of the middle meningeal artery from a temporal bone fracture, though 15–25% are venous in origin.
The classic clinical history is a lucid interval — a patient who is briefly unconscious, wakes up and talks, then deteriorates rapidly into coma as the hematoma expands. Not all EDH patients have a lucid interval, and its absence does not rule out EDH. EDH is a neurosurgical emergency; the outcome with prompt evacuation is generally good because the underlying brain is often uninjured. Mortality rises sharply with every hour of delay once herniation signs appear.
Subdural Hematoma (SDH)
A subdural hematoma is a collection between the dura and the arachnoid membrane, caused by tearing of bridging veins that traverse the subdural space. On CT it appears as a crescent-shaped hyperdense collection that follows the brain surface and can cross suture lines.
- Acute SDH (within 72 hours): bright (hyperdense) on CT; associated with high-energy mechanisms; underlying brain injury is common and worsens prognosis substantially compared to EDH.
- Subacute SDH (3–21 days): becomes isodense to brain and can be difficult to see on CT without contrast.
- Chronic SDH (beyond 21 days): hypodense (dark) on CT; classically in elderly patients, alcoholics, and those on anticoagulants; can occur from trivial or unremembered trauma; presents with headache, confusion, or progressive hemiparesis over weeks.
Acute SDH with mass effect or neurological deterioration typically requires surgical evacuation. Chronic SDH is often drained via burr holes rather than open craniotomy.
Intracerebral Hemorrhage (ICH) / Contusions
Traumatic contusions are areas of bruised brain with microhemorrhages, most commonly in the frontal and temporal poles where the brain impacts the bony ridges of the skull base. On CT they appear as heterogeneous areas with a "salt-and-pepper" pattern of hemorrhage and edema. Contusions are dynamic — they frequently expand over the first 24–48 hours ("contusion expansion" or "blossoming"), which is why patients with significant contusions on initial CT require repeat imaging within 6 hours and often at 24 hours regardless of clinical status.
Traumatic Subarachnoid Hemorrhage (tSAH)
Blood in the subarachnoid space (between the arachnoid and pia mater) is common after TBI and appears as hyperdense sulci on CT. Traumatic SAH is distinguished from aneurysmal SAH by the clinical context and distribution. It is associated with worse outcomes, likely because it reflects greater shearing forces, and carries a risk of cerebral vasospasm (though lower than aneurysmal SAH).
Diffuse Axonal Injury (DAI) on Imaging
DAI is frequently invisible on standard CT. MRI with susceptibility-weighted imaging (SWI) or gradient-echo sequences detects microhemorrhages at the gray-white junction, corpus callosum, and dorsolateral brainstem — the characteristic locations of axonal shearing. Diffusion tensor imaging (DTI) measures fractional anisotropy of white-matter tracts and can quantify the extent of axonal disruption. In clinical practice, the severity of DAI on advanced MRI correlates well with long-term cognitive outcomes.
Acute Emergency Management
The first hour after severe TBI is dominated by the ABCs of trauma resuscitation and simultaneous neurological assessment. The principle is straightforward: you cannot treat the brain if the patient is in hemorrhagic shock or respiratory arrest.
Airway and Ventilation
Patients with GCS ≤8 generally cannot protect their airway and require endotracheal intubation. Hypoxia (SpO<sub>2</sub> <90%) is a powerful secondary injury driver and must be aggressively prevented. Current Brain Trauma Foundation guidelines recommend maintaining PaO<sub>2</sub> >60 mmHg and SpO<sub>2</sub> ≥95%. The target PaCO<sub>2</sub> is 35–45 mmHg (normal). Prophylactic hyperventilation (PaCO<sub>2</sub> <35) is no longer recommended as a routine measure because cerebral vasoconstriction can worsen ischemia; brief hyperventilation is reserved as a temporizing measure for acute herniation.
Blood Pressure
Hypotension is catastrophically harmful in TBI. A single episode of systolic blood pressure (SBP) <90 mmHg doubles mortality. Current guidelines recommend:
- SBP ≥100 mmHg for patients aged 50–69
- SBP ≥110 mmHg for patients aged 15–49 or >70
The physiology: injured brain loses autoregulation of cerebral blood flow, making perfusion directly dependent on systemic blood pressure. The target is cerebral perfusion pressure (CPP) ≥60–70 mmHg, where CPP = mean arterial pressure (MAP) − ICP. Isotonic crystalloids (normal saline) or vasopressors (norepinephrine) are used to maintain MAP; hypotonic fluids (free water, 0.45% saline) are avoided because they worsen cerebral edema.
Initial Neurological Assessment
The GCS is assessed as quickly as possible and should be re-checked frequently because clinical deterioration may be the only sign of hematoma expansion. Key additional assessments include:
- Pupil exam: a fixed and dilated pupil ipsilateral to a mass lesion indicates uncal herniation compressing cranial nerve III — a neurosurgical emergency requiring immediate intervention.
- Limb motor exam: asymmetric weakness or posturing (decorticate or decerebrate) suggests structural injury.
- Vital signs: the Cushing reflex (hypertension + bradycardia + irregular breathing) is a late and ominous sign of severely elevated ICP.
Emergency CT and Triage
Non-contrast CT of the head should be obtained as rapidly as possible in all patients with moderate-to-severe TBI. CT of the cervical spine is obtained simultaneously in most cases because TBI and cervical spine injury frequently coexist. The CT guides the immediate decision: does this patient need the operating room now, or does the ICU for medical management?
Reversal of Anticoagulation
Patients on anticoagulants (warfarin, direct oral anticoagulants, heparin) who sustain intracranial hemorrhage require urgent anticoagulation reversal. For warfarin: 4-factor prothrombin complex concentrate (PCC) plus vitamin K. For direct oral anticoagulants: specific reversal agents (idarucizumab for dabigatran; andexanet alfa for factor Xa inhibitors) if available, or PCC. Platelet transfusion is considered for profound thrombocytopenia.
Intracranial Pressure Monitoring and Control
The skull is a rigid box of fixed volume. When brain tissue, blood, or edema fluid expand within it, intracranial pressure rises. Normal ICP is <15 mmHg. Sustained ICP >20–25 mmHg reduces cerebral perfusion and, if untreated, causes brain herniation and death.
ICP Monitoring Devices
- External ventricular drain (EVD): a catheter placed into the lateral ventricle through a burr hole; it simultaneously measures ICP and can drain cerebrospinal fluid (CSF) to reduce pressure. Gold standard for severe TBI with risk of hydrocephalus or large mass lesions.
- Intraparenchymal monitor (e.g., Camino, Codman): a fiberoptic probe placed directly into brain tissue; measures ICP continuously but cannot drain CSF. Used when EVD placement is technically difficult or contraindicated.
The Brain Trauma Foundation (4th edition guidelines) recommend ICP monitoring for all salvageable severe TBI patients (GCS 3–8 after resuscitation) with abnormal CT findings, or with two or more of: age >40, unilateral or bilateral motor posturing, SBP <90 mmHg.
Stepwise ICP Management Protocol
Management of elevated ICP follows a tiered approach; higher tiers carry greater risk and are escalated sequentially:
- Tier 1 (universal): Head of bed 30°, head midline (optimize venous drainage); normothermia; avoid hypoxia and hypotension; adequate sedation and analgesia; EVD CSF drainage if in place.
- Tier 2 (medical):
- Hyperosmolar therapy: Mannitol (0.25–1.0 g/kg IV bolus) draws water from edematous brain into the bloodstream via osmotic gradient; must monitor serum osmolality (<320 mOsm/kg) and renal function. Hypertonic saline (3% or 23.4% NaCl bolus) is an alternative or adjunct; raises serum sodium and osmolality to draw brain water; may be preferred when hypotension is a concern (unlike mannitol, it expands intravascular volume).
- Short-term hyperventilation: targets PaCO<sub>2</sub> 30–35 mmHg; causes cerebral vasoconstriction and rapidly reduces ICP but at the cost of reduced cerebral blood flow; used as a bridge to definitive treatment.
- Tier 3 (refractory ICP):
- Barbiturate coma (high-dose pentobarbital): dramatically reduces cerebral metabolic demand and ICP; requires continuous EEG monitoring for burst suppression; significant cardiovascular depression; reserved for medically refractory cases.
- Decompressive craniectomy: surgical removal of a large bone flap to allow the swollen brain to expand outward rather than downward through the tentorium. The DECRA trial (2011) showed improved ICP control but worse outcomes in some measures; the RESCUEicp trial (2016) demonstrated a mortality benefit with a corresponding increase in severe disability. Patient selection is critical.
- Therapeutic hypothermia: mild hypothermia (33–35°C) reduces cerebral metabolic rate and ICP; clinical trials have not consistently shown outcome benefit; not currently recommended as routine therapy for ICP control but used in selected cases.
Neurosurgical Interventions
Not all TBI patients require surgery, but rapid identification of those who do is life-saving. The fundamental question is: is there a mass lesion that can be safely evacuated to relieve pressure on the brain?
Craniotomy for Hematoma Evacuation
Open craniotomy with hematoma evacuation is indicated for:
- Epidural hematoma: any EDH >30 mL volume, or >15 mm thickness, or producing >5 mm midline shift, or any EDH with pupil abnormality — regardless of GCS. When caught early, outcomes are excellent.
- Acute subdural hematoma: SDH >10 mm thick or producing >5 mm midline shift, regardless of GCS. SDH with GCS ≤8, GCS drop of ≥2 points, asymmetric or fixed pupils, ICP >20 mmHg unresponsive to medical management. Prognosis is worse than EDH because underlying parenchymal injury is common.
- Traumatic intracerebral hemorrhage: patients with progressive neurological deterioration, medically refractory elevated ICP, or hemorrhage >50 mL may benefit from surgical evacuation, though evidence is less robust than for extra-axial hematomas.
Burr Holes
For chronic subdural hematomas that have liquefied, bilateral burr holes with irrigation and drain placement are usually sufficient and less morbid than open craniotomy. Twist-drill craniostomy under local anesthesia is an option for patients too unstable for general anesthesia.
Decompressive Craniectomy
As discussed under ICP management, decompressive craniectomy involves removing a large section of the skull (typically 12–14 cm diameter) and opening the dura to give the swollen brain room to expand. It is performed either as a primary measure (at the time of hematoma evacuation if brain swelling is severe) or as a secondary measure for refractory ICP. The skull flap is preserved in a freezer or subcutaneously in the patient's abdomen and replaced (cranioplasty) after months, once brain swelling has resolved.
Penetrating TBI
Gunshot wounds and other penetrating injuries to the brain have unique surgical considerations: removal of bone fragments and accessible foreign material is standard, but aggressive debridement deep in eloquent brain tissue risks creating additional deficit. Outcomes for gunshot wounds are poor overall, with in-hospital mortality exceeding 50% for self-inflicted injuries; victims reaching the emergency department alive with GCS >8 have substantially better outcomes.
ICU Care and Systemic Management
Neurocritical care for severe TBI extends well beyond ICP management. The brain is exquisitely sensitive to systemic perturbations, and each of the following must be rigorously controlled.
Temperature Management
Fever (body temperature >38.3°C) dramatically increases cerebral metabolic rate and worsens secondary injury. Normothermia (36–37°C) is the standard target, maintained with antipyretics and, if necessary, surface or intravascular cooling devices. The etiology of fever (infection, drug reaction, central hyperthermia) should always be investigated and treated.
Glucose Management
Both hyperglycemia and hypoglycemia worsen outcomes in TBI. Hyperglycemia (>10 mmol/L / 180 mg/dL) amplifies oxidative stress and inflammation and is independently associated with worse outcomes. Hypoglycemia is immediately harmful to the already-energy-deprived brain. Tight glucose control (4.4–6.1 mmol/L) carries a risk of hypoglycemia episodes; most protocols target a moderate range of 5–10 mmol/L in the ICU.
Seizure Prophylaxis
Post-traumatic seizures occur in approximately 12% of severe TBI patients within the first week (early seizures) and can cause dangerous ICP spikes. Phenytoin (or levetiracetam, which is better tolerated) is recommended for 7 days after severe TBI to prevent early post-traumatic seizures. There is no evidence that prolonged prophylaxis prevents late epilepsy (seizures occurring after 1 week), so prophylaxis is not routinely continued beyond 7 days unless seizures occur. Continuous EEG monitoring is increasingly used to detect non-convulsive status epilepticus, which can be clinically silent in comatose patients.
Coagulopathy
TBI releases massive amounts of tissue factor from the injured brain, triggering a systemic coagulopathy in up to 30% of severe TBI patients. This trauma-induced coagulopathy (TIC) can worsen intracranial hemorrhage and must be identified and corrected rapidly. Serial coagulation studies (PT, PTT, INR, fibrinogen, thromboelastography) guide replacement with fresh-frozen plasma, cryoprecipitate, and platelets.
Nutrition
Severe TBI is a hypermetabolic state; energy expenditure may increase 120–140% above baseline. Early enteral nutrition (within 24–72 hours via nasogastric tube) is associated with lower infection rates and better outcomes. Parenteral nutrition is used when the gut cannot be used. Protein requirements are high (1.5–2.5 g/kg/day) to support healing and prevent muscle wasting.
Deep Vein Thrombosis Prophylaxis
Immobile TBI patients are at high risk for DVT and pulmonary embolism. Mechanical prophylaxis (sequential compression devices) is started immediately. Low-molecular-weight heparin is initiated once intracranial bleeding has been stable on imaging for 24–48 hours, balancing thrombosis and hemorrhage risk.
Ventilator-Associated Pneumonia Prevention
Intubated TBI patients are highly susceptible to ventilator-associated pneumonia (VAP), which worsens outcomes by increasing fever, ICP spikes, and length of ICU stay. Prevention bundles include: head-of-bed elevation to 30–45°, daily sedation vacations and spontaneous breathing trials when ICP allows, oral decontamination, and subglottic secretion drainage.
Rehabilitation and Long-Term Outcomes
Survival from severe TBI is only the first step. The brain has substantial plasticity, particularly in the weeks to months after injury, and early, intensive rehabilitation significantly improves outcomes. Delay or absence of rehabilitation leads to contractures, deconditioning, cognitive stagnation, and depression.
Disorders of Consciousness
Some patients emerge from coma into a vegetative state (eyes open, sleep-wake cycles present, no evidence of awareness) or a minimally conscious state (inconsistent but reproducible evidence of awareness — tracking, command-following, or purposeful movement). These states are frequently misdiagnosed at the bedside; specialized assessments including functional MRI and EEG-based paradigms can detect covert awareness in patients who appear behaviorally unresponsive. The distinction matters enormously for prognosis and treatment decisions.
Cognitive Rehabilitation
Cognitive impairments after moderate-to-severe TBI commonly include attention and concentration deficits, memory impairment, slowed processing speed, executive dysfunction (planning, organization, impulse control), and difficulty with language. Cognitive rehabilitation uses structured, goal-directed tasks to retrain these functions. Evidence supports strategy-based interventions for memory and attention; computer-based cognitive training shows modest benefits as an adjunct.
Physical Rehabilitation
Motor deficits — hemiplegia, balance impairment, gait dysfunction — are addressed by physical and occupational therapy. Early mobilization reduces complications and stimulates neuroplasticity. Spasticity management (baclofen, botulinum toxin) may be needed. Constraint-induced movement therapy and robotic-assisted gait training are evidence-based options for chronic motor deficits.
Behavioral and Psychiatric Sequelae
Depression affects 25–50% of TBI survivors. Anxiety, PTSD, irritability, and emotional dysregulation are also common. Frontal lobe damage disinhibits behavior, producing impulsivity that strains relationships and employment. Treatment combines pharmacotherapy (SSRIs for depression/anxiety; mood stabilizers for irritability; stimulants for cognitive slowing) and behavioral therapies. Family education and caregiver support are essential components of TBI rehabilitation programs.
Chronic TBI and Neurodegeneration
Moderate-to-severe TBI is a significant risk factor for later development of Alzheimer's disease, Parkinson's disease, and other neurodegenerative conditions. The risk is proportional to injury severity and number of injuries. Repetitive mild TBI carries risk for chronic traumatic encephalopathy (CTE) (see Concussion). Long-term neurological follow-up is important for TBI survivors, particularly in the context of cognitive complaints decades after injury.
Prognosis
Outcome after TBI is notoriously difficult to predict in the acute phase. Tools such as the IMPACT (International Mission on Prognosis and Clinical Trial Design in TBI) prognostic model use age, GCS motor score, pupil reactivity, CT findings, and laboratory values to estimate outcome at 6 months. Key points:
- GCS ≤5 with bilateral fixed dilated pupils has a very high mortality rate.
- Age is the strongest single prognostic factor — older brains have less plasticity and more comorbidity.
- Recovery plateaus are typically reached at 12–18 months, but meaningful improvement can continue for years with continued rehabilitation.
- Approximately 40% of severe TBI survivors have significant disability at 1 year; 40% have moderate disability or good recovery; 20% die or remain vegetative.
- Social support, access to rehabilitation, and absence of substance abuse are among the modifiable factors that most influence long-term outcome.
Prevention
Given the limitations of TBI treatment, prevention offers the greatest population-level impact. The three primary causes — falls, motor vehicle accidents, and violence — are all modifiable.
Falls
- Strength and balance training programs (Otago, tai chi) in older adults reduce fall rates by 20–30%.
- Medication review to minimize polypharmacy, sedatives, and orthostatic hypotension.
- Home safety modifications: grab bars, non-slip mats, improved lighting, removal of trip hazards.
- Vision correction and cataract surgery reduce fall risk in older adults.
Motor Vehicle Accidents
- Seat belts reduce TBI fatality risk by approximately 45%.
- Helmets for motorcyclists, cyclists, and recreational vehicle operators are highly effective for preventing fatal head injury.
- Policies targeting drunk driving, distracted driving, and speeding have measurable population-level effects.
- Modern vehicle safety engineering (airbags, crumple zones, automatic emergency braking) has substantially reduced TBI mortality per crash.
Sports and Recreation
Helmets reduce the risk of skull fracture and severe TBI in sports but do not fully prevent concussion. Rule changes (targeting penalties, checking restrictions, heading limits in youth soccer) and better referee enforcement of illegal contact reduce cumulative head-impact exposure.
Violence Prevention
Assault is the third-leading cause of TBI, disproportionately affecting young men in urban settings. Community violence-interruption programs, firearm storage requirements, and domestic violence interventions address the social determinants of TBI at a population level.
Neuroprotective Agents — Current Status
Despite decades of research and dozens of Phase 3 trials, no neuroprotective pharmacological agent has proven effective in TBI at the population level. Progesterone, magnesium, vitamin E, erythropoietin, cyclosporine, and many others have failed to translate from animal models to human benefit. Likely reasons include: the heterogeneity of TBI pathology, inadequate targeting of specific secondary injury mechanisms, and insufficient time resolution between injury and treatment. Ongoing trials are evaluating TXA (tranexamic acid), stem cell therapies, and targeted metabolic interventions.
Research Papers
The following PubMed topic searches return current peer-reviewed literature relevant to moderate-to-severe TBI. Each link opens a live PubMed query.
- Severe traumatic brain injury management
- Intracranial pressure monitoring TBI
- Epidural hematoma surgical outcomes
- Acute subdural hematoma craniotomy
- Decompressive craniectomy TBI
- Hyperosmolar therapy mannitol hypertonic saline
- Diffuse axonal injury MRI DTI
- Secondary brain injury excitotoxicity
- TBI rehabilitation outcomes
- Post-traumatic epilepsy seizure prophylaxis
- Brain Trauma Foundation guidelines
- Minimally conscious state disorders of consciousness
Connections
- Concussion (mTBI)
- Epilepsy
- Stroke
- Alzheimer’s Disease
- Parkinson’s Disease
- PTSD
- Depression
- Anxiety
- Headache
- Brain Fog
- Fatigue
- Magnesium
- Omega-3 Fatty Acids
- Methylene Blue
- Anti-Inflammatory Diet