Choline, Cardiovascular Health & the TMAO Controversy
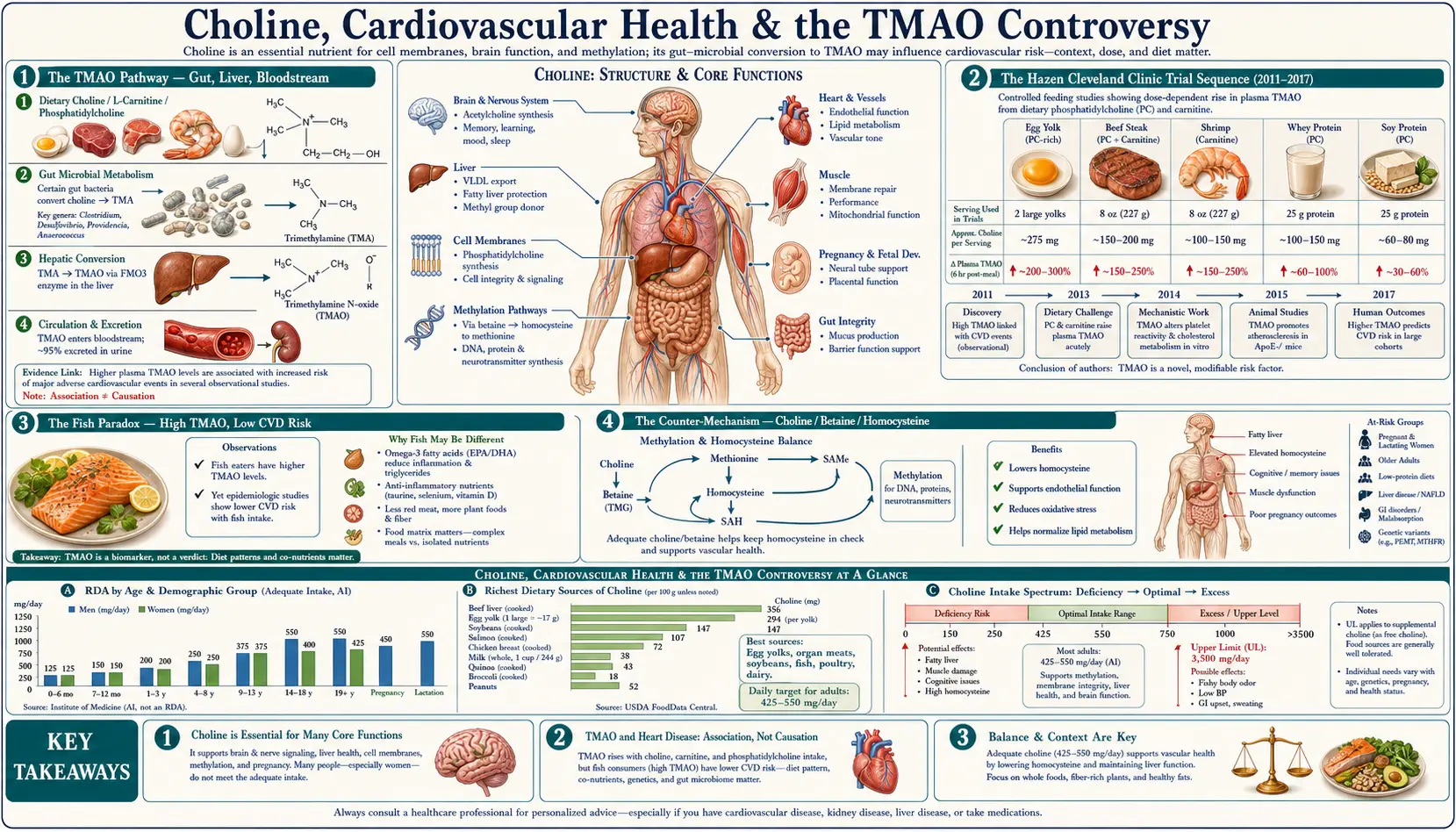
The TMAO controversy is the most contentious topic in choline nutrition. Stanley Hazen's Cleveland Clinic team proposed in 2011 (Nature) and 2013 (NEJM) that gut bacteria convert dietary choline to trimethylamine (TMA), which the liver then oxidizes to trimethylamine N-oxide (TMAO), and that elevated plasma TMAO accelerates atherosclerosis. The mechanistic story is plausible, and the observational data show TMAO-CVD associations. But the epidemiology is conflicting: fish — especially deep-sea fish — produces the highest plasma TMAO elevations of any food, yet fish consumption is associated with REDUCED cardiovascular risk. Choline-rich populations who get their choline from eggs and meat (and who have healthy gut microbiomes) don't obviously have higher CVD than choline-deficient populations. This deep-dive walks through both sides of the debate, the methylation-and-homocysteine axis that probably outweighs the TMAO signal in choline-replete individuals, and the practical guidance for cardiovascular-cautious patients.
Table of Contents
- The TMAO Pathway — Gut, Liver, Bloodstream
- The Hazen Cleveland Clinic Trial Sequence (2011-2017)
- The Fish Paradox — High TMAO, Low CVD Risk
- The Counter-Mechanism — Choline / Betaine / Homocysteine
- What the Choline / CVD Meta-Analyses Actually Show
- The Gut Microbiome Matters More Than Choline Intake
- Choline Deficiency Worsens Dyslipidemia (the Forgotten Mechanism)
- Why TMAO Is Probably a Marker, Not a Cause
- Practical Protocol for the Cardiovascular-Cautious Patient
- Cautions Specific to Cardiovascular Use
- Key Research Papers
- Connections
- Featured Videos
The TMAO Pathway — Gut, Liver, Bloodstream
Trimethylamine N-oxide (TMAO) is produced via a two-organism, two-organ pathway:
- Dietary precursor — choline (in eggs, liver, meat, fish), phosphatidylcholine (in lecithin supplements and food), L-carnitine (in red meat), and betaine (in beets, spinach, wheat germ) all contain or generate trimethylamine moieties.
- Gut bacterial conversion — specific bacterial taxa in the colon (most notably members of the Firmicutes phylum, including Anaerococcus hydrogenalis, Clostridium asparagiforme, Edwardsiella tarda, Streptococcus sanguinis, and others) express the choline TMA-lyase (CutC/CutD) and L-carnitine TMA-lyase (CntA/CntB) enzymes that cleave the trimethylamine moiety from dietary precursors. The free trimethylamine (TMA) is absorbed across the colonic mucosa into the portal circulation.
- Hepatic oxidation — the liver enzyme flavin-containing monooxygenase 3 (FMO3) oxidizes TMA to TMAO. FMO3 expression varies between individuals; women have higher FMO3 expression than men (regulated by estrogen). The rare genetic loss-of-function of FMO3 causes the inability to oxidize TMA, leading to trimethylaminuria ("fish odor syndrome").
- Circulation — TMAO enters systemic circulation, where it can be measured in plasma. Normal fasting plasma TMAO is typically 1-5 µM; postprandial levels rise into the 10-50 µM range after a high-precursor meal; some individuals run chronically elevated above 10 µM.
- Renal excretion — TMAO is freely filtered and excreted in urine. Chronic kidney disease patients have dramatically elevated TMAO levels due to impaired excretion (and this is one of the cleanest contexts in which the TMAO-cardiovascular association may matter clinically).
The key insight: dietary choline does NOT automatically equal TMAO. The conversion depends on the gut microbiome composition. Two people eating identical choline intakes can have 5-10x different plasma TMAO levels depending on which bacterial species dominate their colon. This is why the choline-CVD epidemiology is so messy: the dietary exposure is poorly correlated with the proposed mechanistic biomarker.
The Hazen Cleveland Clinic Trial Sequence (2011-2017)
Stanley Hazen and the Cleveland Clinic Lerner Research Institute group have been the principal investigators on the TMAO-cardiovascular hypothesis. Their work proceeded in steps:
Wang et al. (2011) Nature
The foundational paper. Used metabolomics to identify TMAO as a circulating metabolite predictive of cardiovascular events in a prospective cohort. Showed that germ-free mice given phosphatidylcholine did not develop atherosclerosis (no gut bacteria to make TMA), but conventional mice did. Established the gut-bacteria-as-mediator hypothesis.
Tang et al. (2013) NEJM
4,007 patients undergoing elective coronary angiography. Higher TMAO levels were associated with major adverse cardiac events (death, MI, stroke) over 3-year follow-up, even after adjustment for traditional risk factors. The relative risk in the highest TMAO quartile versus lowest was approximately 2.5.
Koeth et al. (2013) Nature Medicine
Demonstrated that L-carnitine (abundant in red meat) is also a TMA-producing dietary substrate. Showed that long-term meat-eaters had higher TMA-producing bacterial capacity than vegetarians, and that vegetarians given a steak had blunted TMAO responses (because their bacteria didn't have the conversion machinery).
Zhu et al. (2016) Cell
Showed that TMAO directly enhances platelet hyperreactivity and thrombosis risk in animal models. This was an important mechanistic step from "TMAO is a marker" toward "TMAO is a cause."
Senthong et al. (2016) JAMA
Confirmed TMAO as a prognostic marker in patients with peripheral arterial disease.
Through this sequence, Hazen's group built a coherent case for TMAO as both a marker and a contributor to cardiovascular risk. The mechanistic studies have been replicated; the observational associations have been replicated; the platelet-hyperactivity work has been replicated. The case is strong on its own terms.
What the Hazen work has NOT done is run a randomized intervention trial reducing dietary TMAO precursors (or blocking TMA-lyase pharmacologically) and showing that cardiovascular events drop. That would be the clean causal test, and it has not been performed at adequate scale or duration to settle the debate.
The Fish Paradox — High TMAO, Low CVD Risk
The single biggest dietary disruption to the TMAO-causes-CVD story is fish. Deep-sea fish — especially codfish, halibut, and other gadiform species — contain large amounts of pre-formed TMAO (it is an osmolyte that helps deep-sea fish survive high pressure). A single serving of cod can elevate plasma TMAO 10-50x above baseline within hours.
And yet:
- Fish consumption is consistently associated with REDUCED cardiovascular mortality in epidemiological studies (Mozaffarian, Hu, Iso, Bouzan)
- The AHA recommends 2+ servings of fish per week specifically for cardiovascular protection
- The American Mediterranean and Japanese-style traditional diets — both high in fish, both high in TMAO — have among the lowest cardiovascular mortality in the world
- Fish-eating populations don't show the elevated cardiovascular event rates that the TMAO hypothesis would predict
The standard explanation is that fish provides multiple compounds (EPA, DHA, taurine, vitamin D, selenium) that powerfully protect against cardiovascular disease, and that these effects outweigh whatever TMAO is doing. That is plausible. But it raises the question: if fish-induced TMAO is being "overwhelmed" by fish's other benefits, why should we worry about a smaller, less dramatic TMAO elevation from eggs or red meat?
A second possibility: TMAO from fish is biologically different from TMAO from gut-bacterial conversion. Same molecule, but different kinetics, different tissue distribution, different downstream effects. This has not been definitively proved or disproved.
A third possibility: TMAO is correlated with cardiovascular events but is not causally driving them. The TMAO-CVD association may reflect underlying renal dysfunction (CKD patients have very high TMAO; CKD itself is a powerful CVD risk factor), or underlying gut dysbiosis (which has many CVD-relevant consequences beyond TMAO), or underlying dietary patterns that we haven't adequately controlled for.
The fish paradox is, in the considered view of many researchers, a strong argument that the TMAO-CVD relationship is more nuanced than "TMAO is bad, lower it."
The Counter-Mechanism — Choline / Betaine / Homocysteine
Even if we accept some TMAO-mediated cardiovascular risk from dietary choline, there is a powerful competing mechanism running in the OPPOSITE direction: choline supports homocysteine disposal through the betaine-homocysteine methyltransferase (BHMT) pathway. Elevated homocysteine is itself a substantial cardiovascular risk factor (endothelial dysfunction, accelerated atherosclerosis, increased thrombotic risk).
The pathway:
- Choline is oxidized in the liver and kidneys to betaine (trimethylglycine)
- Betaine serves as a methyl donor for the BHMT enzyme, which converts homocysteine back to methionine
- This pathway operates in parallel with the folate-dependent methionine synthase pathway
- When folate or B12 intake is marginal, the choline-betaine pathway becomes the primary route of homocysteine disposal
The clinical implication: choline DEFICIENCY raises homocysteine; choline REPLETION lowers homocysteine. The cardiovascular benefit of normalizing homocysteine likely outweighs any TMAO cost from the same dietary choline in most individuals.
Olthof et al. (2003) demonstrated this directly: supplementation with phosphatidylcholine lowered plasma homocysteine by approximately 10-15% in healthy adults. This is a meaningful cardiovascular biomarker movement. Wallace et al. (2014) showed in NHANES data that low choline intake correlates with elevated homocysteine independent of folate / B12 status.
The competing mechanisms are not symmetrical. The TMAO mechanism applies disproportionately to people with TMA-producing bacteria, with intact FMO3, and presumably with some underlying vascular pathology that makes them sensitive to TMAO's thrombotic effects. The choline-betaine-homocysteine mechanism applies to everyone whose methylation cycle depends on adequate substrate — which is the entire population, but particularly those with MTHFR polymorphisms (~40-45% of population).
What the Choline / CVD Meta-Analyses Actually Show
If TMAO drives cardiovascular risk in proportion to dietary choline intake, you would expect epidemiological studies to consistently show: higher choline intake = higher cardiovascular event rates. What the meta-analyses actually show is more equivocal:
- Meyer & Shea (2017) systematic review of dietary choline / betaine and CVD: no consistent association between higher choline intake and increased CVD events. Some studies showed inverse associations (more choline = lower events), some showed null associations, a few showed weak positive associations.
- Nagata et al. (2015) Japanese cohort of 16,000 women: highest quintile of choline intake had LOWER risk of CHD mortality than lowest quintile.
- Bidulescu et al. (2007) ARIC cohort: no significant association between choline intake and ischemic heart disease or stroke.
- Dalmeijer et al. (2008) Dutch cohort: marginal trend toward INVERSE association between choline + betaine intake and CVD.
- Mygind et al. (2019) Danish cohort: highest tertile of choline intake had lower all-cause mortality.
The pattern: at the epidemiological level, dietary choline does NOT show the consistent positive association with cardiovascular events that the TMAO hypothesis would predict. If anything, the signal is null or weakly inverse.
The most defensible interpretation: TMAO may be a real cardiovascular risk factor in some contexts (CKD patients, individuals with specific microbiome compositions, individuals with underlying vascular pathology), but at the population level, dietary choline within reasonable intake ranges does NOT translate to detectable cardiovascular harm. The competing benefits (homocysteine lowering, hepatic protection, cell membrane integrity, cholinergic neurotransmission) likely outweigh whatever TMAO cost there is.
The Gut Microbiome Matters More Than Choline Intake
If TMAO is the relevant intermediate, then the variable that matters is gut microbiome composition — specifically, whether your colon harbors TMA-producing bacteria in significant abundance. This is partly modifiable:
- Vegetarians and vegans have lower abundance of TMA-producing bacteria (because long-term low choline / carnitine intake selects against them). A vegetarian who eats a steak produces less TMAO from it than a long-term omnivore eating the same steak.
- High dietary fiber shifts microbiome composition toward Bacteroidetes and away from TMA-producing Firmicutes
- Mediterranean diet (high fiber, polyphenols, fermented foods, olive oil) is associated with lower TMAO production capacity
- Probiotics and fermented foods may displace TMA-producing taxa, though the evidence is preliminary
- Antibiotics transiently lower TMAO production capacity (and have been used as a research tool to demonstrate the gut-bacterial mediation)
The practical implication: if you are concerned about TMAO from dietary choline, the highest-impact intervention is probably gut microbiome modification (diet, fiber, fermented foods) rather than choline restriction. Choline restriction has many downsides (cognitive, hepatic, methylation, prenatal); gut microbiome optimization has many upsides beyond just TMAO.
Some clinicians use plasma TMAO testing (available through Cleveland HeartLab and a few other commercial sources) to identify patients who are high TMAO producers. For those individuals, more aggressive gut microbiome intervention and selection of choline forms that are predominantly upper-GI-absorbed (Alpha-GPC, CDP-choline) over those that reach the colon (bitartrate, lecithin) is reasonable.
Choline Deficiency Worsens Dyslipidemia (the Forgotten Mechanism)
An under-appreciated cardiovascular dimension: choline DEFICIENCY directly worsens dyslipidemia through the VLDL mechanism described in the Liver & NAFLD deep dive. Without adequate choline / phosphatidylcholine, the liver cannot properly export triglycerides as VLDL, leading to:
- Hepatic triglyceride accumulation (NAFLD)
- Disturbed plasma lipid profile (elevated triglycerides, sometimes elevated apoB)
- The atherogenic lipid pattern associated with metabolic syndrome
- NAFLD itself is now recognized as an independent cardiovascular risk factor
So the choline-cardiovascular relationship is not unidirectional. Adequate choline:
- Lowers homocysteine (cardioprotective)
- Supports VLDL assembly and prevents NAFLD-driven dyslipidemia (cardioprotective)
- Provides phosphatidylcholine for membrane integrity in vascular endothelium (cardioprotective)
- BUT — in susceptible individuals with TMA-producing microbiomes — raises TMAO (possibly cardiotoxic)
The net effect for any individual depends on their methylation status, their lipid profile, their microbiome composition, their renal function, and their underlying vascular health. For most healthy individuals eating a varied diet, the net effect of adequate choline intake is almost certainly cardioprotective.
Why TMAO Is Probably a Marker, Not a Cause
Several lines of evidence suggest that TMAO is more likely a marker of cardiovascular risk than a direct cause:
- Renal function confounding — TMAO is renally excreted; CKD patients have very high TMAO; CKD itself is a powerful CVD risk factor. Much of the TMAO-CVD association may simply reflect impaired renal clearance in patients who already have cardiovascular disease.
- The fish paradox — fish raises TMAO most dramatically but is cardioprotective.
- The null choline-intake-CVD epidemiology — dietary choline intake doesn't consistently predict CVD events, even though it should if TMAO is the mediator.
- The intervention gap — no large RCT has shown that lowering TMAO (by any means) reduces cardiovascular events. The mechanistic story is plausible; the clinical proof is absent.
- Confounding by underlying gut dysbiosis — TMAO-producing microbiomes are correlated with other adverse microbiome features (inflammation, increased intestinal permeability, metabolic endotoxemia) that have their own CVD implications. TMAO may simply be one measurable marker of an unhealthy gut ecosystem.
The minority view (held by Hazen and collaborators) is that TMAO is causal and that interventions should target the gut bacterial CutC/CutD enzyme or the FMO3 hepatic conversion step. Several small-molecule TMA-lyase inhibitors are in early development. Clinical proof remains pending.
The mainstream view, particularly in cardiology and integrative medicine, is more measured: TMAO is an interesting biomarker that may help stratify cardiovascular risk in some patients, but it does not justify avoidance of choline-rich foods or restriction of choline supplementation in patients who would otherwise benefit (NAFLD patients, pregnant women, cognitively impaired patients).
Practical Protocol for the Cardiovascular-Cautious Patient
For the patient with established CVD or significant cardiovascular risk factors
- Aim for the choline AI (550 mg male / 425 mg female) but no more, primarily from food sources
- Choose fish-based choline sources (salmon, sardines) which come with the EPA / DHA / vitamin D / selenium package
- Optimize gut microbiome: high-fiber diet, fermented foods, Mediterranean pattern
- If supplementing, prefer Alpha-GPC or CDP-choline (upper-GI absorbed) over bitartrate or lecithin (which reach the colon and feed TMA producers)
- Optimize folate, B12, and B6 status to keep the folate-dependent methylation pathway working alongside the choline-betaine pathway
- Check homocysteine; if elevated, addressing it is more important than worrying about TMAO
For the patient with CKD
- Tighter caution is warranted — CKD elevates TMAO independently and CKD is the cleanest context for TMAO-cardiovascular concern
- Discuss with nephrology before significant choline supplementation
- Dietary choline from fish and eggs in moderation is reasonable; high-dose lecithin or carnitine supplementation is not
For the patient with elevated TMAO on lab testing
- Address gut microbiome first (fiber, Mediterranean diet, fermented foods)
- Address any underlying renal dysfunction
- If choline supplementation is needed for other indications (cognitive, hepatic, prenatal), use Alpha-GPC or CDP-choline preferentially
- Recheck TMAO in 3-6 months after intervention
For the otherwise-healthy patient taking choline for cognitive or hepatic indications
- The TMAO concern is real but small relative to the benefits in your demographic
- Proceed with the protocol from the relevant deep dive (Cognition or Liver)
- Optimize gut microbiome as good practice rather than out of TMAO panic
- Don't avoid eggs or fish on TMAO grounds; the evidence doesn't support that level of dietary restriction
Cautions Specific to Cardiovascular Use
- Don't panic over TMAO — the evidence for dietary choline restriction as a cardiovascular intervention is weak. The evidence for benefit of adequate choline intake is strong (homocysteine, hepatic, cognitive). Net likely benefit in most patients.
- CKD changes the calculus — impaired renal clearance plus elevated TMAO plus already-high CVD risk shifts the risk-benefit. Consult nephrology.
- Carnitine in the same conversation — L-carnitine (and acetyl-L-carnitine) are also TMA precursors and have been part of the same controversy. Heavy meat-eaters who supplement with carnitine should consider the same microbiome / form-selection nuances.
- Anticoagulation — the proposed TMAO mechanism includes platelet activation. Patients on anticoagulants or with active thrombotic disease may be a population where caution is warranted. Discuss with cardiology.
- Trimethylaminuria (TMAU) — rare; FMO3 deficiency prevents oxidation of TMA to TMAO, leaving TMA itself to circulate and produce the fish-odor phenotype. Choline restriction is part of TMAU management.
- FMO3 gene variants — common partial-loss-of-function variants affect TMAO levels. Consumer genetic testing may reveal these.
- Sex differences — women have higher FMO3 expression than men (estrogen-driven), so women tend to oxidize TMA to TMAO more efficiently. The clinical implication is unclear.
Key Research Papers
- Wang Z et al. (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. — PubMed 21475195
- Tang WHW et al. (2013). Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. NEJM. — PubMed 23614584
- Koeth RA et al. (2013). Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nature Medicine. — PubMed: Koeth carnitine TMAO
- Zhu W et al. (2016). Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. — PubMed: Zhu TMAO platelet
- Meyer KA & Shea JW (2017). Dietary choline and betaine and risk of CVD: a systematic review and meta-analysis. Nutrients. — PubMed: Meyer Shea choline CVD review
- Nagata C et al. (2015). Choline and betaine intakes are not associated with cardiovascular disease mortality risk in Japanese men and women. Journal of Nutrition. — PubMed: Nagata choline Japanese cohort
- Olthof MR et al. (2003). Low dose betaine supplementation leads to immediate and long-term lowering of plasma homocysteine in healthy men and women. Journal of Nutrition. — PubMed: Olthof betaine homocysteine
- Bidulescu A et al. (2007). Usual choline and betaine dietary intake and incident coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) study. BMC Cardiovascular Disorders. — PubMed: Bidulescu ARIC choline
- Landfald B et al. (2017). Microbial trimethylamine-N-oxide as a disease marker: something fishy? Microbial Ecology in Health and Disease. — PubMed: Landfald TMAO fishy
- Cho CE & Caudill MA (2017). Trimethylamine-N-oxide: friend, foe, or simply caught in the cross-fire? Trends in Endocrinology and Metabolism. — PubMed: Cho Caudill TMAO friend foe
- Senthong V et al. (2016). Plasma TMAO is associated with cardiovascular events in stable atherosclerotic disease. JAMA. — PubMed: Senthong TMAO PAD
- Mygind ND et al. (2019). Dietary betaine and choline intake and mortality risk in Danish cohort. — PubMed: Mygind Danish cohort
PubMed Topic Searches
- PubMed: TMAO and cardiovascular disease
- PubMed: choline / homocysteine / betaine / BHMT
- PubMed: gut microbiome TMA-lyase CutC/CutD
- PubMed: fish intake CVD TMAO paradox
- PubMed: FMO3 and trimethylamine oxidation
Connections
- Choline Overview
- Choline Benefits Hub
- Choline for Cognition
- Choline for Liver & NAFLD
- Choline for Pregnancy
- Cardiovascular Disease
- Homocysteine
- Folate (B9)
- Vitamin B12
- Vitamin B6
- Methionine
- L-Carnitine
- Probiotics
- Fermented Foods
- Nephrology / CKD
- NAFLD
- Salmon
- Eggs