Potassium for Muscle Function
Skeletal muscle contains roughly 70% of the body's total potassium — a staggering 2,800–3,000 mEq stored inside muscle fibers at a concentration approximately 35× the extracellular level. Every voluntary contraction depends on a precise dance between sodium flooding in and potassium flooding out, governed by voltage-gated channels and reset between contractions by the Na+/K+-ATPase pump that consumes roughly 20% of resting energy expenditure in muscle. Hypokalemia produces a textbook progression from leg cramps and weakness through flaccid paralysis and rhabdomyolysis; hyperkalemia produces paresthesias and ascending paralysis with cardiac arrhythmia as the lethal complication. Exercise itself releases potassium from contracting fibers fast enough to transiently elevate plasma K+ to 6–8 mEq/L during maximal effort — the Adolph 1947 hyperkalemia papers documented this phenomenon for the first time. This page maps the molecular machinery, the clinical syndromes at both extremes, the specific cramp and weakness presentations of common potassium-wasting conditions, the exercise-and-recovery cycle, and the dangerous refeeding hypokalemia that kills underweight patients restarted on carbohydrate too aggressively.
Interactive Visualization The Sodium–Potassium Pump — why you have a voltage Run the pump that gives every cell its charge — three sodium out, two potassium in, on Mg-ATP — then drop the magnesium and see exactly why low potassium refuses to correct. Launch →
Table of Contents
- The Na+/K+-ATPase Pump and Resting Potential
- Skeletal Muscle Action Potential
- Hypokalemic Weakness, Cramps, and Paralysis
- Hyperkalemic Paralysis and Paresthesias
- Exercise-Induced Potassium Shifts (Adolph 1947)
- Leg Cramps and Nocturnal Spasms
- Hypokalemic and Hyperkalemic Periodic Paralysis
- Dietary Potassium for Athletes
- Refeeding Hypokalemia
- Smooth Muscle and the GI Tract
- Key Research Papers
- Connections
- Featured Videos
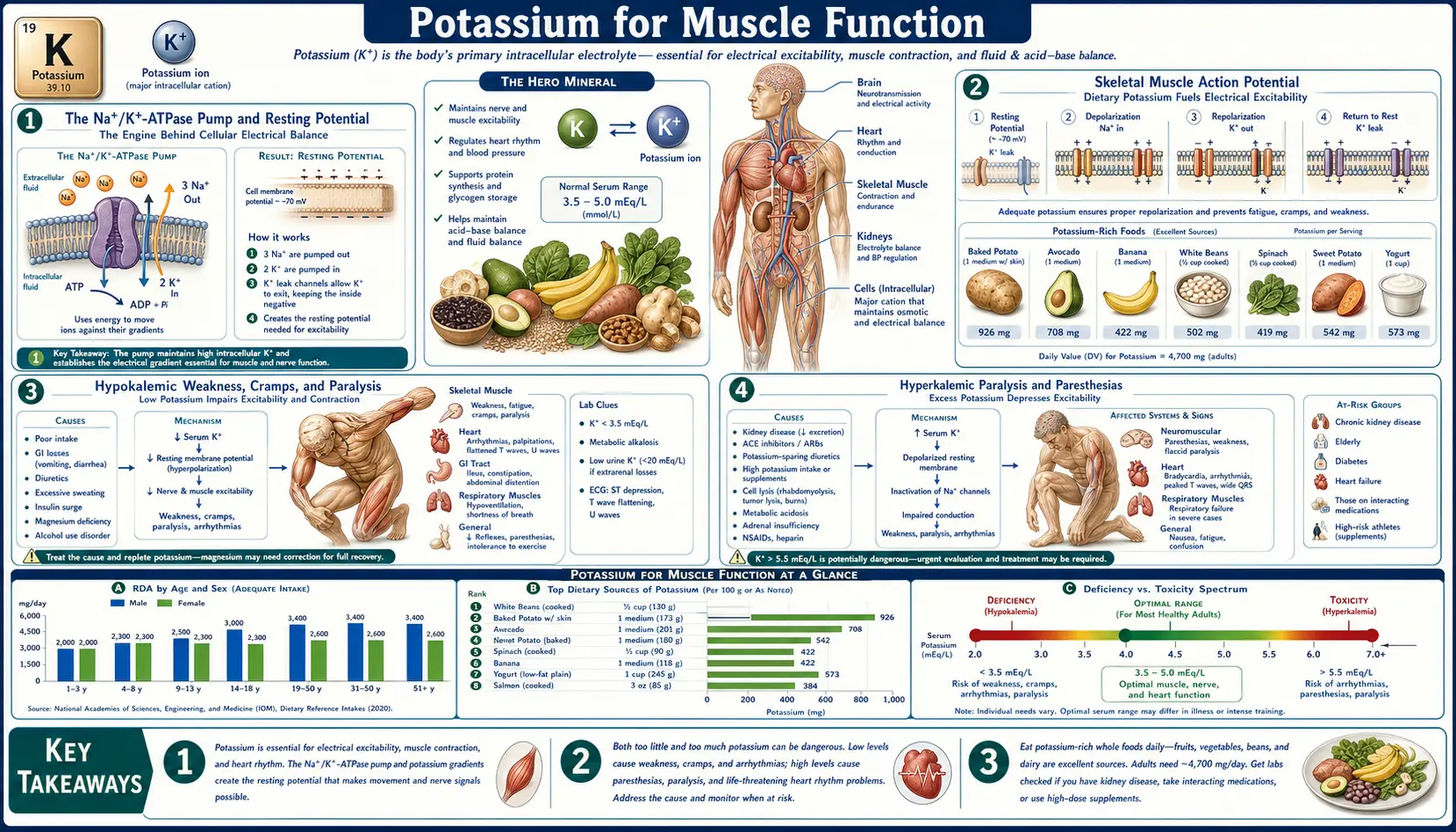
The Na+/K+-ATPase Pump and Resting Potential
Every skeletal muscle fiber maintains a steep electrochemical gradient: intracellular potassium sits at approximately 140–150 mEq/L while the extracellular fluid carries only 3.5–5.0 mEq/L. Inverted relative to that, intracellular sodium is approximately 10 mEq/L against an extracellular concentration of 140 mEq/L. This 35× potassium gradient and 14× sodium gradient are established and maintained by the sodium-potassium ATPase pump (Na+/K+-ATPase), a membrane-spanning enzyme that for every one molecule of ATP hydrolyzed transports three sodium ions out of the fiber and two potassium ions in.
The energy cost is enormous. In resting skeletal muscle the Na+/K+-ATPase accounts for approximately 20% of resting energy expenditure. During intense exercise that figure can climb to 30% or more as the pump works overtime to restore the sodium and potassium displacements caused by each action potential. Across the whole body, basal pump activity consumes roughly 19–28% of basal metabolic rate — a substantial fraction of why warm-blooded mammals require so much food.
The pump establishes the resting membrane potential of approximately −90 mV in skeletal muscle fibers, driven primarily by the potassium gradient through potassium-selective leak channels that allow potassium to slowly diffuse out of the cell down its concentration gradient. The Nernst equation predicts that with normal intracellular and extracellular potassium concentrations, the equilibrium potential for K+ should be approximately −94 mV; the actual resting potential is slightly less negative due to small contributions from sodium and chloride permeability. This polarized resting state is what allows the fiber to fire an action potential the moment a motor neuron releases acetylcholine at the neuromuscular junction.
Skeletal Muscle Action Potential
When acetylcholine binds the nicotinic receptors at the neuromuscular junction, sodium rushes through the receptor channel and depolarizes the post-junctional membrane to threshold. Voltage-gated sodium channels (Nav1.4 in skeletal muscle) snap open, producing a rapid depolarization spike to approximately +40 mV. Within 1–2 milliseconds the sodium channels inactivate and voltage-gated potassium channels open, allowing potassium to flow out of the fiber down its concentration gradient. This potassium efflux drives the membrane potential back toward the resting value — repolarization — resetting the fiber for the next action potential.
Each action potential displaces a small but measurable amount of potassium from inside the fiber to the extracellular fluid. During sustained contractions or repeated firing, this potassium loss accumulates. The local extracellular potassium concentration in the T-tubule system (the invagination of the surface membrane that delivers depolarization into the muscle fiber interior) can rise to 10–15 mEq/L during intense activity — high enough to depolarize the resting potential and slow further sodium channel availability. This is one of the cellular mechanisms underlying muscle fatigue during high-intensity exercise.
Recovery requires the Na+/K+-ATPase pump to actively transport accumulated extracellular potassium back into the fiber while pushing out the sodium that entered during depolarization. The pump's activity is stimulated by both elevated intracellular sodium (its substrate) and by hormonal signals including insulin and catecholamines, which is why the muscular weakness of hypokalemia is reversed within minutes of intravenous potassium administration but only if the pump itself is functioning normally.
Hypokalemic Weakness, Cramps, and Paralysis
Hypokalemia (serum potassium <3.5 mEq/L) produces a remarkably stereotyped clinical syndrome in skeletal muscle. The presentation progresses with worsening severity:
- Mild (3.0–3.5 mEq/L) – Vague fatigue, mild proximal muscle weakness, occasional leg cramps especially at night. Often dismissed as "tired legs" or attributed to age or deconditioning.
- Moderate (2.5–3.0 mEq/L) – Pronounced weakness with difficulty climbing stairs or rising from a chair, painful cramps, twitching (fasciculations), reduced deep tendon reflexes, constipation from smooth-muscle hypotonia.
- Severe (<2.5 mEq/L) – Flaccid paralysis often starting in the lower extremities and ascending; respiratory muscle weakness can produce hypoventilation and ultimately respiratory failure. Ileus develops as gastrointestinal smooth muscle ceases peristalsis.
- Critical (<2.0 mEq/L) – Rhabdomyolysis becomes likely. Severe hypokalemia impairs the local vasodilator response to exercise in working muscle, producing ischemia and muscle fiber breakdown. Creatine kinase rises into the thousands or tens of thousands; myoglobin spills into urine producing the cola-colored color characteristic of rhabdo; acute kidney injury can follow as myoglobin precipitates in renal tubules.
The paradox of hypokalemic weakness is that the resting membrane potential becomes more negative (hyperpolarized) by the Nernst equation, yet sodium channels become functionally less available. The mechanism is that potassium itself regulates sodium channel availability through processes including slow inactivation gating — severe hypokalemia leaves sodium channels in a state where they cannot recover normal availability. Action potential propagation slows, the safety factor for neuromuscular transmission falls, and the fiber simply fails to contract on command.
Common causes of hypokalemia producing muscle symptoms include thiazide and loop diuretic therapy, chronic vomiting or diarrhea, hyperaldosteronism (Conn's syndrome, Cushing's syndrome, licorice ingestion), magnesium depletion (which prevents the kidney from conserving potassium even in the face of low serum levels), and the surreptitious laxative or diuretic abuse seen in some eating disorders.
Hyperkalemic Paralysis and Paresthesias
Hyperkalemia (serum potassium >5.0 mEq/L) produces a partly opposite muscle syndrome. Mild elevation initially increases nerve and muscle excitability because the resting membrane is partially depolarized closer to threshold, which can produce paresthesias (tingling, numbness), muscle twitches, and increased deep tendon reflexes. As potassium rises further the partial depolarization persists long enough to inactivate sodium channels, dropping the safety factor for action potential propagation and producing a weakness progressing to paralysis qualitatively similar to severe hypokalemic paralysis.
- Mild (5.5–6.0 mEq/L) – Paresthesias of the lips and extremities, muscle twitching, mild weakness, often asymptomatic.
- Moderate (6.0–7.0 mEq/L) – Progressive weakness starting in the legs, decreased deep tendon reflexes, palpitations from cardiac involvement.
- Severe (>7.0 mEq/L) – Flaccid ascending paralysis (mimicking Guillain-Barré), respiratory muscle weakness. The cardiac complications — widened QRS, sinusoidal rhythm, ventricular fibrillation — typically kill the patient before they exhaust skeletal muscle function. See the Heart Rhythm page for the cardiac findings in detail.
The clinical importance of recognizing hyperkalemic muscle symptoms early is that the same intervention (calcium gluconate to stabilize the membrane, insulin and glucose to shift potassium into cells, definitive removal of potassium from the body) addresses both the muscle and the cardiac toxicity. Common causes include renal failure (especially with concurrent ACE inhibitor, ARB, or potassium-sparing diuretic use), rhabdomyolysis (releasing intracellular potassium into the circulation), severe tissue trauma or burns, and tumor lysis syndrome.
Exercise-Induced Potassium Shifts (Adolph 1947)
The most counterintuitive feature of muscle potassium handling is that intense exercise produces transient, sometimes substantial, hyperkalemia — not hypokalemia as one might expect from a "depleting" activity. The classic documentation of this phenomenon comes from the work of Edward F. Adolph and colleagues at the University of Rochester in 1947, who measured plasma potassium during and after exhaustive exercise in human volunteers and demonstrated that plasma potassium routinely rose to 6–8 mEq/L during maximal effort, returning to baseline within minutes of cessation.
The mechanism is straightforward: every action potential in a contracting fiber releases potassium into the interstitial space, where it diffuses into the circulation. With many fibers firing many times per second across many large muscle groups during whole-body exercise, the cumulative potassium efflux is enormous — on the order of milliequivalents per minute. The Na+/K+-ATPase pump is being driven hard by both elevated intracellular sodium and by exercise-induced catecholamine release, but it cannot reabsorb potassium faster than active contraction is releasing it.
This exercise hyperkalemia has several physiological consequences:
- Local vasodilation – Elevated interstitial potassium directly activates inward rectifier potassium channels on vascular smooth muscle, hyperpolarizing the cells and producing vasodilation. This is one of several mechanisms (alongside adenosine, nitric oxide, and lactate) that match blood flow to working muscle demand — "active hyperemia."
- Stimulation of ventilation – Plasma potassium stimulates the carotid bodies, contributing to the hyperventilation of intense exercise. This is partly why your breathing rate rises faster than CO2 production during sprinting.
- Cardiac effects – The same hyperkalemia is sensed at the SA node and can blunt the chronotropic response to catecholamines, modestly limiting maximum heart rate. In a patient with marginal cardiac function, exercise-induced hyperkalemia can precipitate arrhythmia — one of the mechanisms behind exertional sudden cardiac death in some athletes.
- Rapid post-exercise normalization – Within 2–5 minutes of stopping exercise, the Na+/K+-ATPase, no longer being out-paced by active efflux, rapidly drives plasma potassium back to baseline. The post-exercise nadir can briefly dip below baseline (mild rebound hypokalemia) before re-equilibrating — clinically irrelevant in healthy people, but a concern in someone on digoxin or with marginal cardiac reserve.
The Adolph 1947 work established that exercise potassium dynamics are completely different from the slow, hormone-mediated potassium balance of resting metabolism — a key insight that informs modern exercise physiology and the management of athletes with potassium-related cardiac risk.
Leg Cramps and Nocturnal Spasms
The garden-variety leg cramp — a sudden, painful, involuntary contraction usually in the calf, often at night, lasting seconds to minutes — is commonly attributed to potassium deficiency in popular health writing. The clinical reality is more nuanced. Nocturnal leg cramps in healthy adults are most often associated with one of the following:
- Magnesium deficiency — arguably more common than frank hypokalemia, and clinically more responsive to repletion. Magnesium is required as a cofactor for the Na+/K+-ATPase, so magnesium depletion produces a functional intracellular potassium deficiency even when serum potassium appears normal. See the Cramp Prevention and Magnesium Replenishment pages.
- Dehydration and electrolyte imbalance — particularly after heavy sweating, athletic activity, or diuretic use. Sodium and chloride losses interact with potassium and magnesium balance.
- Muscle overuse and altered mechanics — new exercise, unusually long walks, prolonged standing, or footwear changes can produce localized cramping unrelated to systemic electrolyte status.
- True hypokalemia — usually only in the context of diuretic use, chronic vomiting/diarrhea, or eating-disorder behaviors. In these settings, cramping is one of several findings and a serum potassium check is the next step.
For most healthy adults with nocturnal cramps, the practical approach is: (1) check magnesium status, (2) ensure adequate hydration, (3) increase potassium intake from whole foods (see the Rich-Foods page), (4) consider stretching the affected muscle before bed. Routine potassium supplementation is rarely the answer and can produce hyperkalemia in older adults with reduced renal reserve or those on RAAS-blocking medications.
Exercise-associated cramping — the calf or hamstring cramp that strikes a runner mid-race or a basketball player in the fourth quarter — is a separate syndrome whose dominant mechanism is now believed to be altered neuromuscular control (Schwellnus' "altered neuromuscular control" hypothesis) rather than electrolyte depletion. The traditional electrolyte-replacement-only approach addresses one of several contributing factors but not the dominant mechanism in most athletes.
Hypokalemic and Hyperkalemic Periodic Paralysis
The periodic paralysis syndromes are a family of rare inherited disorders in which a defect in a muscle ion channel produces intermittent attacks of flaccid weakness or paralysis triggered by shifts in plasma potassium. They are genetically distinct conditions that share an electrophysiological theme:
- Hypokalemic Periodic Paralysis (HypoKPP) — an autosomal-dominant condition caused most commonly by mutations in the CACNA1S gene (encoding the dihydropyridine receptor / Cav1.1 calcium channel) or SCN4A (sodium channel). Attacks of severe flaccid weakness or paralysis lasting hours are triggered by carbohydrate-rich meals (which drive insulin-mediated potassium uptake into cells), by rest after vigorous exercise, by stress, or by salt loading. Serum potassium during an attack can drop to 1.5–2.5 mEq/L. Treatment is with potassium supplementation during attacks, dietary potassium-loading and carbohydrate-restriction for prevention, and carbonic anhydrase inhibitors (acetazolamide, dichlorphenamide) as prophylaxis. A secondary thyrotoxic periodic paralysis with identical clinical features occurs in some Asian patients with hyperthyroidism.
- Hyperkalemic Periodic Paralysis (HyperKPP) — an autosomal-dominant condition caused by mutations in SCN4A (sodium channel) that prevent normal sodium channel inactivation in response to modest depolarization. Attacks of weakness or paralysis are triggered by rest after exercise, by fasting, by cold exposure, or by potassium-rich meals. Serum potassium during an attack rises to 5.5–7.0 mEq/L. Treatment is with carbohydrate ingestion or intravenous glucose-insulin during attacks, dietary potassium-restriction for prevention, and thiazide diuretics or carbonic anhydrase inhibitors as prophylaxis. Patients often develop a chronic myopathy with fixed weakness later in life.
- Andersen-Tawil Syndrome — an autosomal-dominant channelopathy caused by KCNJ2 mutations affecting the Kir2.1 inward rectifier potassium channel. The triad is periodic paralysis (can present as either hypo- or hyperkalemic), cardiac arrhythmias (long QT, ventricular ectopy), and characteristic dysmorphic features.
These are rare conditions but worth recognizing because of the unique trigger pattern and because the inappropriate intervention can worsen attacks (giving potassium during a HyperKPP attack, or giving glucose-insulin during a HypoKPP attack).
Dietary Potassium for Athletes
The athletic population has somewhat different potassium considerations than the general population, but the key insight is that the body's potassium homeostasis is robust enough that well-fed athletes very rarely become hypokalemic from exercise alone. The dominant risk factors for athletic hypokalemia are concurrent issues — eating disorders, diuretic abuse for "making weight" in combat sports or rowing, severe rapid weight cuts, gastroenteritis combined with continued training, or the rare misuse of beta-agonist asthma medications (which drive potassium into cells).
For routine training and recovery the recommendations are:
- Eat potassium-rich whole foods daily — bananas, potatoes, beans, leafy greens, dairy, salmon. A diverse whole-food diet hitting the 3,400 mg/day AI for men or 2,600 mg/day for women is more than enough to support training and recovery in nearly all athletes.
- Sports drinks are over-engineered for potassium — most contain only 30–40 mg of potassium per serving (compared to 400+ mg in a banana). For workouts under 90 minutes water is sufficient; for longer sessions in hot conditions, sodium replacement matters far more than potassium.
- Endurance athletes losing 1–2% bodyweight in sweat per hour may need to consciously plan potassium-rich food intake in the post-workout meal, especially if training twice a day, but supplementation is rarely needed and can produce hyperkalemia if combined with NSAID use that impairs renal potassium handling.
- The post-workout banana remains a sensible practice not because of any acute potassium emergency but because it provides 400+ mg of potassium, easily-absorbed carbohydrate to restore liver and muscle glycogen, and the small magnesium contribution all in one inexpensive package.
- Coconut water contains roughly 600 mg of potassium per cup, more than a banana, with the bonus of being a hypotonic fluid that absorbs faster than plain water. It's a reasonable post-workout rehydration choice, though over-priced for what it provides.
The exception is the athlete with a separately diagnosed potassium-wasting condition (Gitelman syndrome, Bartter syndrome, primary hyperaldosteronism) for whom training amplifies the underlying renal potassium losses and supplementation is genuinely needed.
Refeeding Hypokalemia
One of the most dangerous and under-recognized clinical scenarios involving potassium and muscle is refeeding syndrome, which can kill underweight or chronically malnourished patients when carbohydrates are reintroduced too aggressively. The mechanism involves potassium, phosphate, magnesium, and thiamine all simultaneously, but the potassium component is responsible for much of the cardiac and muscle morbidity.
In starvation, the body adapts to use fat and protein as fuel, depleting intracellular phosphate, magnesium, and potassium stores even while serum concentrations may appear normal due to renal conservation and shifts from intracellular pools. The reintroduction of carbohydrate triggers a surge of insulin release. Insulin drives glucose into cells, and along with the glucose, it drives potassium, phosphate, and magnesium into cells via the Na+/K+-ATPase pump and through phosphorylation of glycolytic intermediates. Serum potassium can drop precipitously from a borderline-normal baseline to dangerous levels within hours.
The clinical presentation includes:
- Severe muscle weakness — including respiratory muscle weakness producing hypoventilation and hypoxia
- Cardiac arrhythmia — the same arrhythmias seen in any severe hypokalemia, including torsades de pointes and ventricular fibrillation
- Heart failure — from low magnesium, low phosphate (which impairs ATP regeneration in cardiac myocytes), and high circulating insulin causing fluid retention
- Wernicke encephalopathy — if thiamine is not replaced before glucose, the surge in glycolysis can precipitate clinically catastrophic thiamine deficiency
- Death — from cardiac arrhythmia, respiratory failure, or both
Populations at highest risk: patients hospitalized after prolonged starvation (anorexia nervosa, severe alcohol use disorder, prolonged NPO status in critical illness, prisoner-of-war survivors), patients on prolonged total parenteral nutrition started too fast, and the elderly post-surgical patient who has been hospitalized for a week without adequate nutrition then started on enteral feeds aggressively.
The clinical management is conservative reintroduction of nutrition (start at 5–10 kcal/kg/day for the highest-risk patients and advance over a week), pre-emptive thiamine before any carbohydrate, daily monitoring and pre-emptive replacement of potassium, phosphate, and magnesium during the first 5–7 days, and continuous cardiac monitoring for those with serum potassium below 3.0 mEq/L. The NICE refeeding syndrome guidelines and the more recent ASPEN/ASPEN consensus criteria provide the operational framework most hospitals follow.
Smooth Muscle and the GI Tract
The same potassium principles that govern skeletal muscle apply, with modifications, to smooth muscle throughout the body. Smooth muscle resting membrane potentials are typically less negative (−55 to −60 mV) than skeletal muscle, and contraction is governed by complex interactions between voltage-gated calcium channels, calcium-activated potassium channels, and second-messenger systems. The most clinically visible consequences of potassium disturbance in smooth muscle are gastrointestinal:
- Hypokalemia produces ileus — the cessation of normal peristaltic activity. Patients with serum potassium <2.5 mEq/L commonly develop abdominal distension, nausea, vomiting, and constipation as smooth muscle tone collapses. This is a well-known complication of severe hypokalemia in the inpatient setting.
- Vascular smooth muscle and blood pressure — covered in detail on the Blood Pressure page. Elevated extracellular potassium hyperpolarizes vascular smooth muscle through Kir channels and stimulates endothelial nitric oxide release, producing vasodilation and lowering peripheral resistance.
- Bronchial smooth muscle — potassium plays a modest role through SK and BK calcium-activated potassium channels that modulate airway tone. Beta-agonist asthma medications (albuterol, salmeterol) drive potassium into cells via beta-2 receptor stimulation of the Na+/K+-ATPase; severe bronchospasm treated with high-dose nebulized albuterol can produce a measurable transient drop in serum potassium of 0.5–1.0 mEq/L, occasionally clinically important in patients on diuretics.
- Bladder smooth muscle — chronic hypokalemia is associated with urinary retention and, with prolonged severity, with the renal-vacuolar nephropathy pattern that can permanently impair urinary concentrating ability.
Key Research Papers
- Adolph EF, Brown AH, Goddard DR, et al. (1947). Physiology of man in the desert — including the classic studies on plasma potassium during exercise. Interscience Publishers, New York. — PubMed
- Clausen T (2003). Na+-K+ pump regulation and skeletal muscle contractility. Physiological Reviews;83(4):1269-1324. — DOI: 10.1152/physrev.00011.2003
- Lindinger MI, Sjøgaard G (1991). Potassium regulation during exercise and recovery. Sports Medicine;11(6):382-401. — PubMed
- Sejersted OM, Sjøgaard G (2000). Dynamics and consequences of potassium shifts in skeletal muscle and heart during exercise. Physiological Reviews;80(4):1411-1481. — DOI: 10.1152/physrev.2000.80.4.1411
- Cannon SC (2010). Voltage-sensor mutations in channelopathies of skeletal muscle. Journal of Physiology;588(11):1887-1895. — DOI: 10.1113/jphysiol.2010.186874
- Schwellnus MP, Drew N, Collins M (2008). Muscle cramping in athletes — risk factors, clinical assessment, and management. Clinics in Sports Medicine;27(1):183-194. — PubMed
- Mehler PS, Brown C (2015). Anorexia nervosa — medical complications. Journal of Eating Disorders;3:11. — DOI: 10.1186/s40337-015-0040-8
- Crook MA, Hally V, Panteli JV (2001). The importance of the refeeding syndrome. Nutrition;17(7-8):632-637. — DOI: 10.1016/S0899-9007(01)00542-1
- Mehanna HM, Moledina J, Travis J (2008). Refeeding syndrome: what it is, and how to prevent and treat it. BMJ;336(7659):1495-1498. — DOI: 10.1136/bmj.a301
- Statland JM, Tawil R (2018). Periodic Paralysis. Continuum (Minneap Minn);24(6):1696-1711. — DOI: 10.1212/CON.0000000000000676
- Maughan RJ, Shirreffs SM (2019). Muscle cramping during exercise: causes, solutions, and questions remaining. Sports Medicine;49(Suppl 2):115-124. — DOI: 10.1007/s40279-019-01162-1
- Wong P, Chen JI, Lyster H (2022). Hypokalemia and rhabdomyolysis. BMJ Case Reports. — PubMed
PubMed Topic Searches
- PubMed — Na/K-ATPase in skeletal muscle and exercise
- PubMed — Hypokalemic periodic paralysis (CACNA1S)
- PubMed — Hyperkalemic periodic paralysis (SCN4A)
- PubMed — Refeeding syndrome hypokalemia
- PubMed — Exercise-associated muscle cramps and electrolytes
Connections
- The Sodium–Potassium Pump — interactive animation
- Potassium Overview
- Hypokalemia (Low Potassium) Symptoms
- Hypokalemia and Muscle Weakness
- Hypokalemia and Muscle Cramps
- Potassium Benefits Hub
- Potassium and Blood Pressure
- Potassium and Heart Rhythm
- Potassium-Rich Foods
- Magnesium (Na/K-ATPase Cofactor)
- Magnesium and Heart Health
- Calcium
- Cramp Prevention
- Magnesium Replenishment
- Arrhythmia
- Kidney Disease
- Heart Palpitations
- Bananas
- Avocado