Relationship Between Hemoglobin and Ceruloplasmin: Why Iron-Deficiency Anemia Is Often Functional Copper Deficiency
The standard story about anemia goes: low hemoglobin means low iron, so take iron. The standard story is incomplete. Iron without ceruloplasmin cannot be loaded into hemoglobin — and ceruloplasmin is a copper-dependent enzyme. When ceruloplasmin runs low, a person can have ample iron stores (or even iron overload) and still present with all the symptoms of anemia: fatigue, pallor, exercise intolerance, and a low MCV or low hemoglobin on a CBC. Treating that picture with isolated iron supplements often makes the fatigue worse, because unbound ferrous iron drives Fenton chemistry, produces hydroxyl radicals, and oxidatively damages tissue while the bone marrow still cannot finish a complete red cell. This page walks through the biochemistry — the ferroxidase mechanism of ceruloplasmin, the hephaestin step at the enterocyte, the chain of copper enzymes red cell precursors depend on — and explains why Morley Robbins' "root-cause copper" framework has gained traction among patients whose iron-only protocols never resolved their symptoms.
Table of Contents
- Overview: The Copper-Iron Connection
- Ceruloplasmin Biology and the Ferroxidase Mechanism
- Why Iron Needs Cu²⁺-Enzymes to Load into Hemoglobin
- Hephaestin and Intestinal Iron Absorption
- Iron-Deficiency Anemia vs Functional Ceruloplasmin Deficiency
- Morley Robbins' Root-Cause Framework
- Serum Copper vs Ceruloplasmin Labs and the Bioavailable Copper Calculation
- Dietary Sources: Beef Liver, Oysters, Dark Chocolate
- Copper-Zinc Ratio and Supplementation Cautions
- Practical Workup and Corrections
- Research Papers and References
- Connections
- Featured Videos
Overview: The Copper-Iron Connection
Iron does not move through the body alone. At every step between the gut and the developing red blood cell, iron has to ride on a carrier protein, hand off across a membrane, or get inserted into a heme ring — and most of those steps require a copper-dependent enzyme to oxidize iron from its ferrous (Fe²⁺) to its ferric (Fe³⁺) state. Without that oxidation:
- Transferrin — the iron-carrying protein in plasma — cannot bind iron at all. Transferrin only binds Fe³⁺.
- Enterocytes (gut lining cells) cannot release dietary iron into the bloodstream.
- Macrophages (which recycle ~25 mg of iron daily from senescent red cells) cannot mobilize the iron they recover.
- Hepatocytes (which store iron as ferritin) cannot export their stores when erythropoiesis needs them.
This is why a patient with normal or high serum ferritin can still present as iron-deficient on a CBC. The iron is there. The copper-dependent oxidative machinery that mobilizes it is not. — Search PubMed reviews this circuitry in detail and frames it as a single integrated copper-iron axis — not two independent mineral systems.
Ceruloplasmin Biology and the Ferroxidase Mechanism
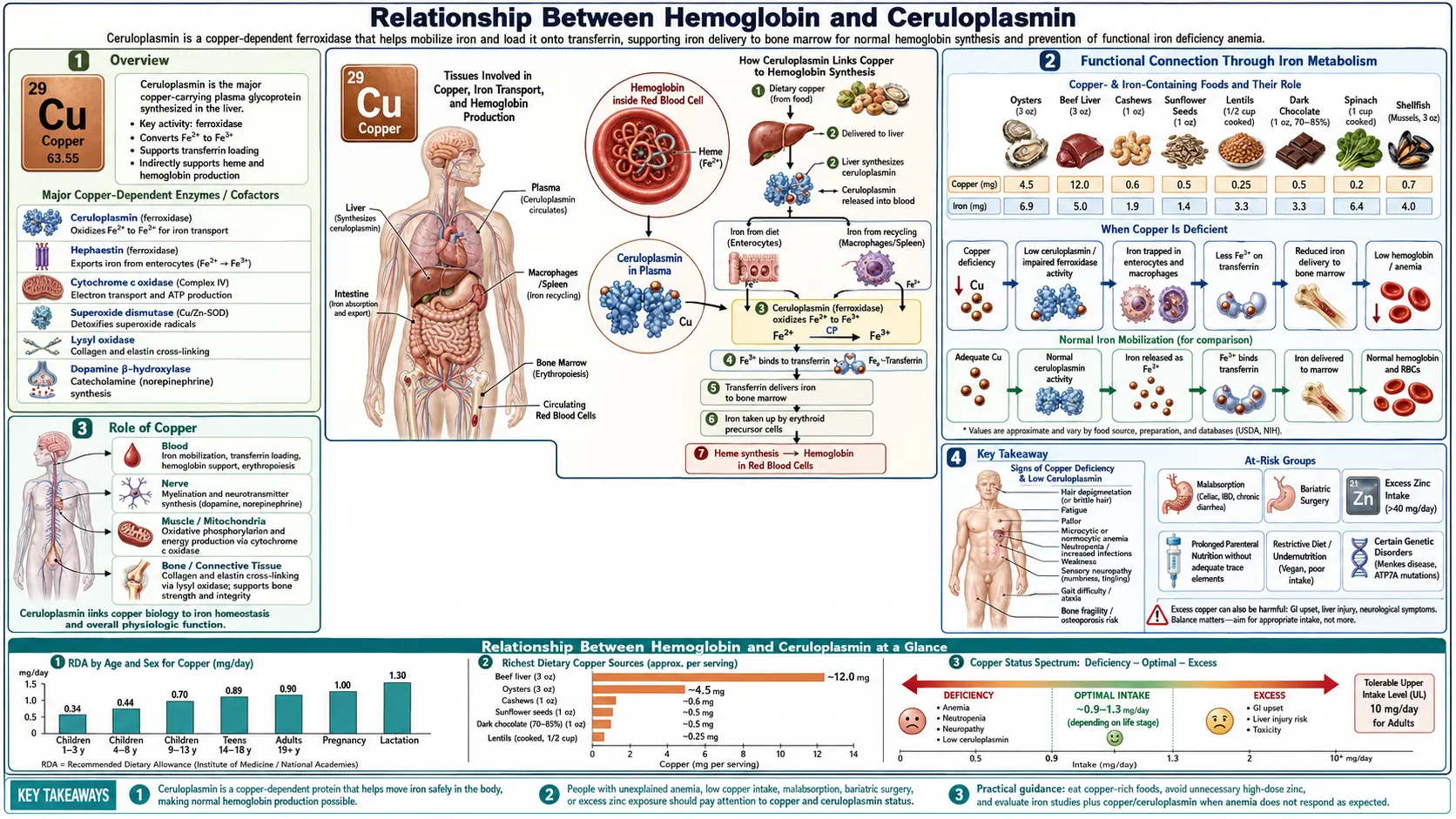
Ceruloplasmin is a 132 kDa glycoprotein synthesized in the liver. Each molecule binds six atoms of copper and accounts for roughly 90–95% of total copper in plasma. (Albumin and ceruloplasmin together carry essentially all the rest. "Free" copper in the loose sense is < 5% of circulating copper.) This is the headline biology: serum copper is mostly ceruloplasmin-bound copper.
The catalytic surface of ceruloplasmin holds a tri-copper cluster (two Type-3 atoms plus one Type-2 atom) plus three additional Type-1 "blue copper" sites. The cluster catalyzes the four-electron reduction of O₂ to H₂O while simultaneously oxidizing four Fe²⁺ substrates to Fe³⁺. That coupled reaction is the ferroxidase reaction, and ceruloplasmin is the main ferroxidase in plasma. — Search PubMed reviewed the molecular biology of ceruloplasmin and the consequences of loss-of-function mutations — a condition called aceruloplasminemia, which produces neurodegeneration with iron accumulation in the brain and liver. Their work made the point starkly: without ceruloplasmin, iron piles up in the wrong places and never reaches the bone marrow.
Three properties of ceruloplasmin matter clinically:
- Copper-loading happens during synthesis. If a patient is copper-deficient, the liver releases apoceruloplasmin — the protein scaffold without its copper atoms. Apoceruloplasmin is enzymatically inactive and degraded within hours. An immunoassay that measures the protein scaffold (the common clinical assay) cannot tell the difference between active and inactive ceruloplasmin.
- It is an acute-phase reactant. Inflammation, infection, pregnancy, and estrogen (including hormonal contraceptives) raise serum ceruloplasmin. A "normal" ceruloplasmin in an inflamed patient may actually be a functional copper deficiency that's only crossing the lower reference limit because of acute-phase upregulation.
- It is the dominant antioxidant ferroxidase in plasma. Beyond loading transferrin, ceruloplasmin clears the Fe²⁺ that leaks into circulation from injured cells — preventing the free iron that would otherwise catalyze hydroxyl-radical formation via Fenton chemistry. — Search PubMed demonstrated that mice lacking the related ferroxidase machinery accumulate iron in tissues and develop progressive oxidative damage.
Why Iron Needs Cu²⁺-Enzymes to Load into Hemoglobin
Picture the journey of a single iron atom from a piece of beef on the plate to an oxygen-carrying heme group inside a mature red blood cell. There are at least four obligatory copper-dependent steps:
- Enterocyte efflux. A duodenal enterocyte absorbs Fe²⁺ via the DMT1 transporter on its apical (gut-facing) membrane. To leave the cell on the basolateral (blood-facing) side, iron has to pass through ferroportin and then be oxidized back to Fe³⁺ by hephaestin, a copper-dependent ferroxidase membrane-bound to the enterocyte. Without hephaestin, ferroportin cannot complete its hand-off, and iron stays trapped inside the gut cell — eventually sloughed off into the lumen and lost.
- Plasma transport. Once iron makes it into the portal blood, it must immediately bind transferrin to avoid causing oxidative damage. Transferrin only binds Fe³⁺. The Fe²⁺ that hephaestin and ceruloplasmin produced is now ready for binding. If oxidation fails, the iron remains as Fe²⁺, binds nothing, and immediately starts catalyzing hydroxyl-radical formation.
- Macrophage recycling. Every day, splenic and hepatic macrophages digest senescent red blood cells and recover roughly 25 mg of iron — about 25 times the daily dietary iron requirement. Recycled iron exits macrophages through ferroportin and is loaded onto transferrin by ceruloplasmin acting as the macrophage-side ferroxidase. This recycling pathway is the actual source of most of the iron that reaches the bone marrow each day. — Search PubMed showed that ceruloplasmin-knockout mice cannot mobilize iron from macrophages and develop iron-loaded reticuloendothelial cells alongside hypoferremic plasma.
- Cytochrome c oxidase and heme assembly. Inside the mitochondrion of an erythroid precursor, the heme ring is built by inserting iron into protoporphyrin IX via ferrochelatase. Heme synthesis is energy-demanding and depends on mitochondrial ATP from oxidative phosphorylation — which itself depends on cytochrome c oxidase (Complex IV), a copper-containing enzyme with two essential copper centers (Cu₀ and Cu₋). A copper-deficient erythroid precursor has impaired mitochondrial ATP production and impaired heme synthesis — even when iron and transferrin are abundant.
These steps are the structural reason why copper deficiency produces an anemia that looks identical to iron-deficiency anemia on routine labs: microcytic or normocytic, hypochromic, low reticulocytes, low serum iron, low transferrin saturation — but with a critical difference. Ferritin and total body iron stores may be normal or elevated. Iron supplementation does not fix it. — Search PubMed documented this pattern in adult patients and emphasized that copper deficiency should be considered in any anemia that does not respond to iron repletion.
Hephaestin and Intestinal Iron Absorption
Hephaestin (Greek for "smith," after the metal-working god Hephaestus — a deliberate naming nod to its iron-handling role) is a copper-dependent ferroxidase discovered in 1999 in the search for the molecular defect underlying sex-linked anemia (sla) mice. The sla mouse cannot absorb iron normally from the gut despite intact DMT1 and ferroportin. — Search PubMed identified the defective gene as a ceruloplasmin homologue tethered to the enterocyte basolateral membrane. The gene was named hephaestin (HEPH).
Hephaestin's role is anatomically critical: ceruloplasmin is a plasma enzyme and cannot reach into the enterocyte cytoplasm. The cell needs its own membrane-bound ferroxidase to oxidize iron immediately as it exits via ferroportin, so that transferrin in the portal blood can bind it. Without hephaestin, iron either backs up inside the enterocyte (eventually shed when the cell turns over) or leaks into portal blood as Fe²⁺ that no carrier protein can grab. — Search PubMed confirmed the obligate coupling of ferroportin and hephaestin in human enterocytes and showed that copper-deficient enterocytes produce inactive apo-hephaestin, mirroring the ceruloplasmin pattern.
Clinically: copper status at the level of the gut epithelium is upstream of iron absorption. A patient with adequate dietary iron and adequate ferroportin can still develop iron-deficient labs if hephaestin is unloaded. This is one mechanism by which chronic low copper intake, high-zinc supplements (which down-regulate enterocyte copper uptake), bariatric surgery, or chronic PPI use can produce iron-resistant anemia.
Iron-Deficiency Anemia vs Functional Ceruloplasmin Deficiency
The two pictures overlap almost completely on a standard CBC and iron panel. The critical distinction is what shows up on labs that most primary-care physicians do not order:
- True iron deficiency: low serum iron, low ferritin (< 30 ng/mL), low transferrin saturation, high TIBC. Responds to oral or IV iron.
- Functional copper/ceruloplasmin deficiency: low serum iron, normal or high ferritin, low or low-normal transferrin saturation, low or low-normal ceruloplasmin, low serum copper, sometimes low neutrophils (copper deficiency causes neutropenia), and a history of either zinc supplementation, gastric bypass, prolonged TPN, or the kind of high-cereal / low-organ-meat / low-shellfish modern diet that delivers < 1 mg copper daily. — Search PubMed reviewed copper-deficiency anemia and emphasized that the simultaneous neutropenia (and, in chronic cases, myelopathy resembling B12 deficiency) is the major clue separating it from iron-deficiency anemia.
The unhelpful clinical reality is that the standard reflex in primary care for any anemia + fatigue presentation is iron, with rare follow-up testing. Patients can spend years on oral iron supplements, watching their ferritin climb into the 200s while their hemoglobin barely budges and their fatigue persists. That iron is being banked in the liver and reticuloendothelial system because ceruloplasmin cannot recycle it back out. The longer this continues, the more oxidative damage accumulates from unbound Fe²⁺.
This is what the literature increasingly calls "functional iron deficiency" — the iron is present but not bioavailable to erythropoiesis. The mainstream framing usually invokes inflammation and hepcidin upregulation as the mechanism (anemia of chronic disease). The copper-ceruloplasmin axis is the same problem at a different choke-point, and the two often coexist in the same patient.
Morley Robbins' Root-Cause Framework
Morley Robbins, a former hospital administrator turned mineral-balance educator, is the leading lay voice translating this biochemistry into a clinical protocol. His core thesis is that the modern epidemic of fatigue, anemia, hypothyroid symptoms, and "low energy" is fundamentally a root-cause copper and magnesium deficiency masquerading as iron and vitamin D deficiencies. The framework, fully developed in his book and Root Cause Protocol, rests on several claims that align with the biochemistry above:
- Bioavailable copper has collapsed in the modern food supply. Soil copper is depleted by industrial farming; organ meats and shellfish (the historical concentrated sources) are no longer staples; glyphosate, which is a copper chelator, has saturated the food supply since the 1990s. Glyphosate copper chelation is one of his frequent emphases.
- Iron supplementation and iron-fortified flour have created a population-wide iron overload that the liver cannot release because ceruloplasmin is undermade. This is the picture documented in Iron Overload as Hidden Toxicity.
- The fix is whole-food copper, not isolated copper supplements. Robbins is firm: copper from beef liver, oysters, and bee pollen comes packaged with the cofactors (especially retinol from animal foods) needed to make functional ceruloplasmin. Cuprous oxide and cupric sulfate — the cheap forms in multivitamins — do not reliably load apoceruloplasmin. See Whole Food Copper Sources.
- Retinol (preformed vitamin A) is the limiting cofactor for ceruloplasmin synthesis. Beta-carotene cannot substitute, because the conversion is impaired in many people. Without retinol, the liver simply cannot make functional ceruloplasmin even if copper is adequate.
- Magnesium is the upstream regulator of the whole copper-iron-energy system. Magnesium runs the enzymes that build ceruloplasmin and that recycle iron through ferritin. Magnesium-depleted patients almost universally have ceruloplasmin issues too. See Magnesium Replenishment.
The framework has critics in mainstream hematology (who point out that frank copper-deficiency anemia is uncommon outside of specific risk groups), but it accurately describes the biochemistry, predicts the lab pattern, and has produced reproducible symptom resolution in patients whose iron-only protocols failed. For the patients sitting at the intersection of "still tired despite ferritin of 200" and "ceruloplasmin never tested," it is a legitimate diagnostic and therapeutic lens.
Serum Copper vs Ceruloplasmin Labs and the Bioavailable Copper Calculation
The lab interpretation here is genuinely tricky, and it is where most clinical workups go off the rails. Two facts to anchor:
- Serum copper measures total copper — ceruloplasmin-bound copper plus albumin-bound copper plus the trace free pool.
- Ceruloplasmin is usually measured immunologically — the assay detects the protein scaffold, not its enzymatic activity. Apoceruloplasmin (copper-empty) and holoceruloplasmin (copper-loaded) both register.
This produces a critical interpretive trap: a patient can have a "normal" ceruloplasmin number by immunoassay while the protein is mostly apo (inactive). The serum copper will also drift low (because most copper is loaded onto the active enzyme). The patient looks normal on both numbers individually but is functionally copper-deficient.
The workaround used in functional medicine and in the Morley Robbins framework is the bioavailable copper calculation (sometimes called "non-ceruloplasmin copper" or the "ceruloplasmin-to-copper ratio"):
- Each milligram of ceruloplasmin holds ~0.3% copper by weight (3 μg Cu per mg ceruloplasmin).
- Multiply serum ceruloplasmin (mg/dL) by 3 to get the μg/dL of copper accounted for by ceruloplasmin.
- Subtract that from total serum copper (μg/dL) to get the non-ceruloplasmin (bioavailable) copper.
- Worked example: serum copper 100 μg/dL, ceruloplasmin 25 mg/dL. Ceruloplasmin accounts for 25 × 3 = 75 μg/dL. Non-ceruloplasmin copper = 100 − 75 = 25 μg/dL.
- Reference range target (Robbins, functional medicine): non-ceruloplasmin copper should be roughly 5–15 μg/dL, and it should be a small fraction (about 10–20%) of total copper. A high fraction — say, > 25 μg/dL or > 25% of total — suggests apoceruloplasmin loading on the assay (unbound copper drifting freely, with the protein not properly loaded).
This calculation is the same math used in clinical hepatology to screen for Wilson disease (an autosomal-recessive disorder of copper export, in which non-ceruloplasmin copper rises far higher because ATP7B can't load copper onto apoceruloplasmin). The functional-medicine application uses the same arithmetic at the milder end of the same spectrum.
For more, see the Ceruloplasmin and Bioavailable Copper page. For ordering bloodwork, see Lab Tests.
Dietary Sources: Beef Liver, Oysters, Dark Chocolate
The single highest-density food source of bioavailable copper, by a wide margin, is beef liver. A 3-oz serving of cooked beef liver delivers roughly 12–14 mg of copper — more than 1000% of the RDA — alongside the retinol, B12, choline, and biotin that the liver uses to build functional ceruloplasmin. Historically liver was a weekly staple in most cultures; the post-1950s shift to muscle-meat-only animal foods has removed almost all dietary copper from the typical Western plate. Robbins and other root-cause-protocol practitioners are unequivocal on this point: one ounce of beef liver daily, or 4–6 oz once or twice weekly, is the foundational copper intervention.
Other concentrated whole-food sources, in approximate descending order of copper density per typical serving:
- Oysters (especially Pacific) — 4–7 mg copper per 6-oyster serving; also rich in zinc, B12, and selenium.
- Beef and lamb liver — the canonical organ source.
- Bee pollen — small but bioavailable copper alongside a broad mineral and B-vitamin profile; one of Robbins' frequently recommended supplements.
- Dark chocolate / cacao — 70%+ cacao chocolate delivers roughly 0.5–1 mg copper per oz; a 1-oz daily piece is a pleasant supplementary source. See Dark Chocolate.
- Spirulina, shiitake mushrooms, nuts (cashews, hazelnuts, Brazil nuts), sunflower seeds, sesame seeds — plant sources; bioavailability is lower than from organ meats because of phytate binding.
- Lentils, chickpeas, and other legumes — meaningful contributions in plant-based diets if combined with adequate retinol from other sources (which is itself the hard part of plant-based diets).
- Beef — muscle meat is a minor copper source (~0.1 mg per 3 oz) but combined with hepcidin-friendly heme iron makes it part of the broader package. See Beef.
Olivares & Uauy 1996 (PMID 8780340) reviewed dietary copper intake in human populations and concluded that frank copper deficiency from diet alone is rare in adults eating mixed diets — but emphasized that subclinical, functional deficiency is plausible in modern Western diets dominated by refined grains and lacking organ meats.
Copper-Zinc Ratio and Supplementation Cautions
Copper and zinc are biological antagonists at the gut epithelium. High-dose zinc supplementation down-regulates copper absorption by upregulating metallothionein in the enterocyte, which preferentially binds copper and prevents its transport into the bloodstream. The classic case report literature includes multiple patients who developed full-blown copper-deficiency anemia and myelopathy after months to years of using zinc-containing denture creams, lozenges, or high-dose zinc supplements for cold prevention. Willis et al. 2005 (PMID 16115980) reported a case series in which zinc-induced copper deficiency produced both anemia and myelopathy reversible only with copper repletion.
The healthy dietary copper-to-zinc ratio sits around 1:8 to 1:12 — roughly 1 mg copper per 8–12 mg zinc. Most multivitamins and most "immune support" zinc lozenges blow this ratio dramatically — a typical zinc lozenge delivers 25–50 mg zinc with zero copper, or a tiny "balance" dose of 1–2 mg copper as cupric oxide (a particularly poorly absorbed form).
Practical guardrails:
- Avoid zinc supplementation > 25 mg/day for more than 2–4 weeks without matching whole-food copper intake.
- Avoid "balanced" multivitamins that contain 50 mg zinc and 2 mg cupric oxide — the cupric oxide form is essentially inert; this configuration is a slow-rolling copper deficiency.
- For copper repletion, the Robbins framework strongly favors food-source copper (beef liver, oysters, bee pollen) over any isolated supplement. The position is that copper packaged with retinol and B-vitamins is the only form that reliably loads apoceruloplasmin.
- If supplementing, copper bisglycinate is generally preferred over cupric oxide, but should be paired with retinol-containing foods (eggs, butter, liver) on the same day for ceruloplasmin assembly.
- Watch for the inverse problem too: chronic copper toxicity (rare, but real in patients on copper IUDs, or on long-term high-dose copper supplements without zinc) presents with anxiety, depression, hair loss, and acne.
Practical Workup and Corrections
For a patient with chronic fatigue, anemia that doesn't respond to iron, or "always tired despite normal labs," the workup that actually answers the copper-iron question is:
- CBC with differential and reticulocyte count. Look for anemia, neutropenia (copper deficiency causes both), and low retics.
- Full iron panel: serum iron, TIBC, transferrin saturation, ferritin. Pay close attention to ferritin: if ferritin is normal or high while hemoglobin is low, iron supplementation is not the answer.
- Serum copper AND ceruloplasmin, ordered together so the bioavailable copper calculation can be done. Most labs measure both. Calculate non-ceruloplasmin copper = total copper − (3 × ceruloplasmin).
- Retinol (serum vitamin A as retinol, not "vitamin A" by carotenoid screen). This is the limiting cofactor for ceruloplasmin synthesis.
- RBC magnesium (not serum magnesium — serum is a poor marker because magnesium is intracellular). Magnesium regulates ceruloplasmin synthesis upstream.
- Inflammatory markers (CRP, ferritin as an acute-phase reactant): inflammation falsely elevates ceruloplasmin. Interpret accordingly.
If the pattern fits functional copper deficiency — low or low-normal ceruloplasmin, low non-ceruloplasmin copper, normal/high ferritin, fatigue that didn't respond to iron — the first-line correction is dietary, not supplemental:
- Beef liver, 1 oz daily or 4 oz twice weekly (fresh or freeze-dried capsules from a clean source).
- Oysters or other shellfish weekly as a copper + zinc + B12 package.
- Cod liver oil or grass-fed beef tallow / butter for retinol cofactor.
- 1 oz of 70%+ dark chocolate daily for sustained low-dose copper.
- Pull any high-dose zinc supplements not paired with copper.
- If supplementing magnesium, prefer glycinate or malate (not oxide); 200–400 mg elemental daily.
- Hold isolated iron supplements until ceruloplasmin status is corrected. Re-test the full panel after 8–12 weeks of dietary repletion.
For patients with frank symptomatic copper deficiency (myelopathy, severe anemia, neutropenia), formal copper repletion under hematology supervision is required — the dietary approach above is for sub-clinical functional deficiency, not for established disease. — Search PubMed reviewed the management of copper-deficiency myelopathy and emphasized that neurological deficits, once established, may only partially reverse even with prolonged repletion — making early identification critical.
Research Papers and References
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
Connections
- All Minerals
- Copper Overview
- Copper Benefits Deep Dive
- Hemoglobin and Ceruloplasmin (Benefits sub-article)
- Iron
- Zinc
- Magnesium
- Morley Robbins (Root Cause Protocol)
- Ceruloplasmin and Bioavailable Copper
- Copper-Iron Dysregulation
- Iron Overload Hidden Toxicity
- Whole Food Copper Sources
- Root Cause Protocol
- Magnesium Replenishment
- Glyphosate Copper Chelation
- Cure Your Fatigue Book
- Anemia
- Fatigue
- Complete Blood Count (CBC)
- Lab Tests
- Beef
- Dark Chocolate
- Vitamin A (Retinol)
- Vitamin C
- Oxidative Stress