Myasthenia Gravis
1. Overview
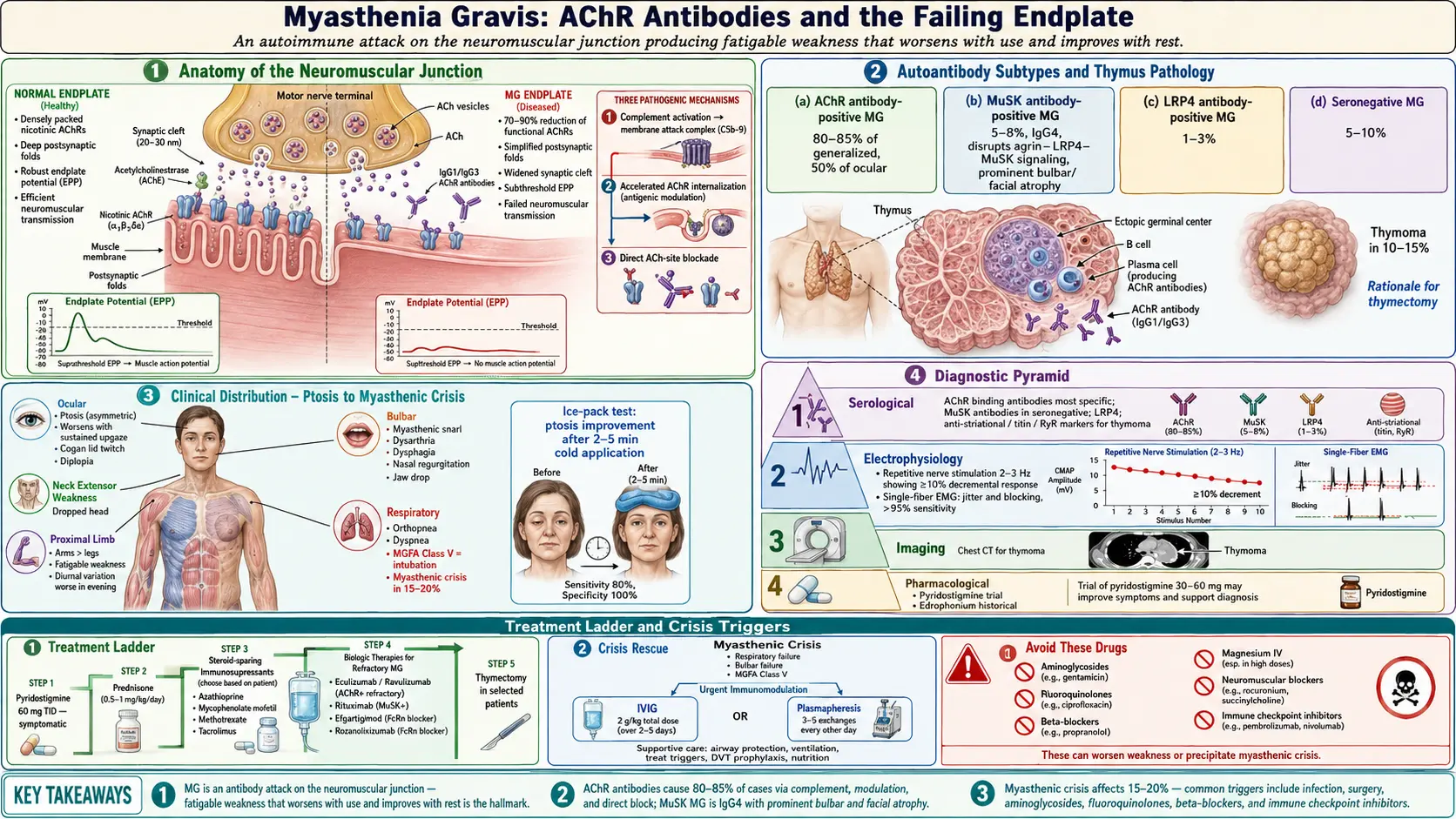
Myasthenia gravis (MG) is a chronic autoimmune neuromuscular disorder characterized by fluctuating weakness and fatigability of voluntary (skeletal) muscles. The hallmark of the disease is muscle weakness that worsens with activity and improves with rest. MG is caused by autoantibodies that target components of the neuromuscular junction (NMJ), most commonly the nicotinic acetylcholine receptor (AChR), disrupting normal neuromuscular transmission.
The name "myasthenia gravis" derives from the Greek and Latin words meaning "grave muscular weakness," reflecting the historically high mortality rate before modern treatments were available. Today, with appropriate therapy, most patients achieve good symptom control and have a near-normal life expectancy. However, myasthenic crisis — acute worsening requiring mechanical ventilation — remains a life-threatening emergency that occurs in approximately 15-20% of patients during their disease course.
Myasthenia gravis is classified by several schemes:
- By autoantibody type:
- AChR antibody-positive MG — approximately 80-85% of generalized MG and 50% of ocular MG
- MuSK (muscle-specific kinase) antibody-positive MG — approximately 5-8% of generalized MG
- LRP4 (low-density lipoprotein receptor-related protein 4) antibody-positive MG — approximately 1-3%
- Seronegative MG — approximately 5-10%; antibodies may be present at levels below detection thresholds
- By clinical distribution (Myasthenia Gravis Foundation of America classification):
- Class I — ocular weakness only (ptosis, diplopia)
- Class II — mild generalized weakness (IIa: predominantly limb/axial; IIb: predominantly oropharyngeal/respiratory)
- Class III — moderate generalized weakness
- Class IV — severe generalized weakness
- Class V — intubation required (myasthenic crisis)
- By thymus pathology:
- Thymoma-associated MG — approximately 10-15% of MG patients have a thymoma
- Thymic hyperplasia-associated MG — approximately 65% of AChR-positive MG patients under 50
- Normal or involuted thymus — more common in late-onset MG
- By age of onset:
- Early-onset MG (EOMG) — onset before age 50; predominantly female; associated with thymic hyperplasia
- Late-onset MG (LOMG) — onset after age 50; increasingly male predominance; associated with thymic atrophy
2. Epidemiology
Myasthenia gravis has an annual incidence of approximately 7-23 per million and a prevalence of 150-300 per million, though recent studies suggest the prevalence may be increasing due to improved diagnosis and longer survival with treatment. In the United States, an estimated 60,000-80,000 individuals are living with MG.
MG exhibits a bimodal age distribution. In younger patients (under age 50), there is a female predominance with a peak incidence in the second and third decades (female-to-male ratio approximately 3:1). In older patients (over age 50), there is a male predominance with a peak incidence in the sixth to eighth decades. The overall incidence of late-onset MG has been increasing over the past several decades, possibly due to better recognition, aging populations, and the use of immune checkpoint inhibitors in cancer therapy.
Neonatal MG occurs in approximately 10-20% of infants born to mothers with MG, caused by transplacental transfer of maternal AChR antibodies. This is typically transient, resolving within 2-3 months as maternal antibodies are cleared. Juvenile MG (onset before age 18) accounts for approximately 10-15% of all MG cases in Western populations but a higher proportion in East Asian populations, where juvenile ocular MG is particularly common. Thymoma-associated MG has a peak incidence in the fourth to sixth decades with no sex predilection.
MG can be associated with other autoimmune diseases in approximately 15-20% of patients, including autoimmune thyroid disease (the most common), rheumatoid arthritis, systemic lupus erythematosus, and type 1 diabetes mellitus.
3. Pathophysiology
The pathophysiology of MG centers on autoimmune attack at the neuromuscular junction, the specialized synapse where motor neurons communicate with skeletal muscle fibers.
Normal Neuromuscular Transmission
When an action potential reaches the motor nerve terminal, voltage-gated calcium channels open, triggering the release of acetylcholine (ACh) vesicles into the synaptic cleft. ACh diffuses across the cleft and binds to nicotinic acetylcholine receptors (AChRs) clustered on the postsynaptic muscle membrane at the endplate. This binding opens cation channels, generating an endplate potential (EPP). When the EPP exceeds threshold, a muscle fiber action potential is initiated, leading to contraction. ACh is rapidly hydrolyzed by acetylcholinesterase (AChE) in the synaptic cleft. The safety factor of neuromuscular transmission is normally large, with EPPs exceeding the threshold by a significant margin.
AChR Antibody-Mediated Pathology
In the most common form of MG, IgG1 and IgG3 antibodies target the AChR and cause damage through three mechanisms:
- Complement-mediated destruction — antibodies activate the classical complement cascade, generating the membrane attack complex (C5b-9), which damages the postsynaptic membrane and destroys AChRs. This is the dominant pathogenic mechanism in AChR-MG
- Accelerated AChR internalization (antigenic modulation) — cross-linking of AChRs by divalent antibodies promotes receptor endocytosis and degradation, reducing the number of functional receptors on the muscle surface
- Direct blockade — some antibodies directly block the ACh binding site on the receptor, preventing neuromuscular transmission
The combined effect is a reduction in functional AChR density by up to 70-90%, simplification of the postsynaptic folds, and widening of the synaptic cleft. This reduces the safety factor of neuromuscular transmission, so that with repeated nerve stimulation, fewer ACh quanta are released (presynaptic depletion), and the diminished EPPs fail to reach threshold, resulting in the characteristic fatigable weakness.
MuSK Antibody-Mediated Pathology
MuSK (muscle-specific kinase) is a transmembrane tyrosine kinase essential for clustering AChRs at the neuromuscular junction during development and maintaining their density in adult muscle. MuSK antibodies are predominantly IgG4, which does not activate complement. Instead, MuSK antibodies inhibit the agrin-LRP4-MuSK signaling pathway, disrupting AChR clustering and reducing endplate AChR density. MuSK-MG has distinct clinical features, including prominent oropharyngeal and respiratory weakness, facial and tongue atrophy, and a less reliable response to cholinesterase inhibitors.
LRP4 Antibody-Mediated Pathology
LRP4 (low-density lipoprotein receptor-related protein 4) is the receptor for agrin, the nerve-derived signal that activates MuSK. LRP4 antibodies block the agrin-LRP4 interaction, disrupting the same AChR clustering pathway as MuSK antibodies. LRP4-MG is generally milder and may have a better prognosis.
Role of the Thymus
The thymus plays a central role in the immunopathogenesis of AChR-MG. In patients with thymic hyperplasia, the thymus contains ectopic germinal centers with B cells that produce AChR antibodies, T helper cells that provide help to these B cells, and myoid cells that express AChR and may serve as the autoantigen source. In thymoma-associated MG, the tumor disrupts normal T cell selection, allowing autoreactive T cells to escape to the periphery. This thymic involvement provides the rationale for thymectomy as a therapeutic intervention.
4. Etiology and Risk Factors
Autoimmune Etiology
MG is an autoimmune disease in which loss of immune tolerance leads to the production of pathogenic autoantibodies against NMJ components. The trigger for this loss of tolerance is unknown in most cases but likely involves a combination of genetic susceptibility and environmental factors.
Genetic Susceptibility
- HLA associations — early-onset MG is strongly associated with HLA-B8, HLA-DR3, and HLA-A1 haplotypes (particularly in Caucasian populations); late-onset MG is associated with HLA-DR2, HLA-B7, and HLA-DRB1*15:01
- Non-HLA genetic factors — polymorphisms in CTLA-4, PTPN22, TNFRSF11A, and IL-2 receptor genes have been associated with MG susceptibility
- Familial MG — rare; approximately 3-5% of patients have a first-degree relative with MG; the concordance rate in identical twins is approximately 35-40%
Environmental Triggers
- Infections — molecular mimicry between microbial antigens and AChR epitopes has been proposed; specific infectious triggers remain unidentified
- Medications — certain drugs can unmask or exacerbate MG:
- Immune checkpoint inhibitors — anti-PD-1 (nivolumab, pembrolizumab), anti-PD-L1 (atezolizumab), and anti-CTLA-4 (ipilimumab) can trigger de novo MG, which is often severe and has high mortality
- D-penicillamine — can induce MG that typically resolves after drug discontinuation
- Interferon-alpha — rarely triggers MG
- Stress, surgery, and infection — can trigger exacerbations in established MG
Risk Factors for Myasthenic Crisis
- Respiratory infections — the most common trigger
- Surgery — particularly thymectomy and procedures requiring general anesthesia
- Medications that impair neuromuscular transmission — aminoglycosides, fluoroquinolones, beta-blockers, magnesium, neuromuscular blocking agents, botulinum toxin
- Rapid tapering of immunosuppression
- Emotional stress
- Pregnancy — especially postpartum
- High environmental temperatures
5. Clinical Presentation
Ocular Symptoms
- Ptosis (drooping eyelid) — the most common initial symptom, occurring in approximately 50% of patients at presentation; often asymmetric; worsens with sustained upward gaze; the Cogan lid twitch sign (transient overshoot of the eyelid upon return from downgaze) is characteristic
- Diplopia (double vision) — results from weakness of extraocular muscles; can mimic any pattern of cranial nerve palsy; fluctuating nature helps distinguish it from structural lesions
- Orbicularis oculi weakness — inability to fully close the eyes; tested by asking the patient to squeeze their eyes shut while the examiner attempts to open them ("peek sign")
- Approximately 50-60% of patients who present with purely ocular symptoms will develop generalized disease within 2 years; if ocular symptoms remain isolated for 2 years, only about 10-15% will subsequently generalize
Bulbar Symptoms
- Dysarthria — nasal, slurred, or breathy speech; may worsen with prolonged talking (fatigable dysarthria)
- Dysphagia — difficulty swallowing, particularly with prolonged meals; nasal regurgitation of liquids; risk of aspiration
- Jaw weakness — difficulty chewing, particularly tough or fibrous foods; jaw may hang open (jaw drop)
- Facial weakness — bilateral facial weakness producing a characteristic "myasthenic snarl" when attempting to smile; transverse smile
- Weak tongue and palate — impaired articulation and food manipulation
- Bulbar symptoms are particularly prominent in MuSK-MG, where facial, lingual, and palatal weakness with atrophy may be striking
Limb and Axial Weakness
- Proximal limb weakness — arms affected more than legs; difficulty with overhead activities (combing hair, reaching shelves), climbing stairs, and rising from chairs
- Neck weakness — inability to hold the head upright ("dropped head syndrome"); neck extensors weaker than flexors
- Fatigable weakness — the hallmark of MG; strength is relatively normal at rest but deteriorates with sustained or repeated effort and recovers after rest
- Diurnal variation — symptoms are typically better in the morning after rest and worsen as the day progresses
Respiratory Symptoms
- Dyspnea — initially on exertion; may progress to respiratory failure
- Orthopnea — diaphragmatic weakness causing difficulty breathing when supine
- Myasthenic crisis — acute respiratory failure requiring intubation and mechanical ventilation; occurs in approximately 15-20% of patients; can be triggered by infection, surgery, medication changes, or may occur spontaneously
Key Examination Findings
- Fatigable weakness — demonstrated by sustained upward gaze (ptosis worsens over 1-2 minutes), repeated deltoid abduction, or sustained arm elevation
- Ice pack test — placing ice on the ptotic eyelid for 2-5 minutes improves ptosis by reducing acetylcholinesterase activity at the cooled NMJ; sensitivity approximately 80%, specificity approximately 100%
- Normal sensory examination — sensation is completely normal in MG; any sensory abnormality should prompt consideration of alternative diagnoses
- Preserved or brisk reflexes — reflexes are normal in MG; reduced reflexes suggest alternative diagnoses
- No muscle atrophy in typical AChR-MG (though atrophy of facial and tongue muscles may occur in MuSK-MG)
6. Diagnosis
Serological Testing (Autoantibodies)
- AChR binding antibodies — the primary diagnostic test; positive in approximately 85% of generalized MG and 50% of ocular MG; highly specific (>99%); false positives are rare but can occur in thymoma without MG and in patients on immune checkpoint inhibitors
- AChR modulating antibodies — measure complement-mediated AChR destruction; increases sensitivity when added to binding antibody assay
- AChR blocking antibodies — measure direct blockade of the ACh binding site; less commonly used
- MuSK antibodies — tested in AChR-negative generalized MG; present in approximately 40-50% of seronegative generalized cases; predominantly IgG4
- LRP4 antibodies — tested in double-seronegative (AChR and MuSK negative) patients; available primarily in research laboratories
- Striational (anti-striated muscle) antibodies — antibodies against titin, ryanodine receptor, and Kv1.4; found in approximately 80-90% of thymoma-associated MG; useful as a marker for thymoma, especially in patients under 50
Electrophysiological Studies
- Repetitive nerve stimulation (RNS) — the standard electrodiagnostic test; motor nerve is stimulated at 2-3 Hz and compound muscle action potentials (CMAPs) are recorded; a decremental response of 10% or more between the first and fourth/fifth CMAP is diagnostic; sensitivity approximately 75% in generalized MG and 30-50% in ocular MG; testing proximal muscles and clinically weak muscles increases sensitivity
- Single-fiber electromyography (SFEMG) — the most sensitive electrodiagnostic test for MG; measures jitter (variability in the time interval between action potentials of two muscle fibers innervated by the same motor neuron) and blocking; sensitivity exceeds 95% in generalized MG and 85-90% in ocular MG; less specific than RNS (jitter can be increased in neuropathies and motor neuron diseases)
Pharmacological Testing
- Edrophonium (Tensilon) test — historically important but less commonly used due to the risk of bradycardia and the availability of serological tests; intravenous edrophonium (a short-acting acetylcholinesterase inhibitor) produces rapid, temporary improvement in weakness; must have an objective endpoint (ptosis, diplopia); atropine must be available for potential cholinergic side effects
- Pyridostigmine trial — a therapeutic trial of oral pyridostigmine (60 mg three times daily) with objective improvement supports the diagnosis
Imaging
- CT or MRI of the chest — mandatory to evaluate for thymoma; CT with contrast is the standard; approximately 10-15% of MG patients have a thymoma; thymoma screening should be performed in all newly diagnosed MG patients
Pulmonary Function Testing
- Forced vital capacity (FVC) — serial measurements are essential for monitoring respiratory function; FVC below 20 mL/kg (or below 1 liter) indicates impending myasthenic crisis; the "20-30-40 rule" guides intubation decisions: FVC below 20 mL/kg, maximal inspiratory pressure (MIP) below -30 cmH2O, or maximal expiratory pressure (MEP) below 40 cmH2O
- Negative inspiratory force (NIF/MIP) — measures diaphragmatic strength; more sensitive than FVC for detecting early respiratory compromise
7. Treatment
MG treatment is individualized based on disease severity, antibody subtype, thymus pathology, age, and comorbidities. The goals are to achieve minimal manifestation status (MMS) or complete stable remission (CSR) while minimizing treatment side effects.
Symptomatic Treatment
- Pyridostigmine (Mestinon) — the first-line symptomatic treatment; an acetylcholinesterase inhibitor that increases ACh availability at the NMJ; typical dose is 60 mg every 4-6 hours (maximum approximately 480 mg/day); provides symptomatic relief but does not alter disease course; cholinergic side effects include abdominal cramping, diarrhea, increased salivation, bradycardia; less effective in MuSK-MG and may worsen symptoms in some MuSK patients
- Extended-release pyridostigmine (Mestinon Timespan) — 180 mg at bedtime for patients with morning weakness
Immunosuppressive Therapy
- Corticosteroids (prednisone) — the most commonly used first-line immunosuppressive agent; provides benefit in approximately 75-80% of patients; initiated at a low dose and gradually increased to avoid transient worsening; long-term use limited by side effects (osteoporosis, diabetes, hypertension, cataracts, weight gain, infections)
- Azathioprine (Imuran) — steroid-sparing agent; onset of action 6-12 months; often combined with prednisone initially; check TPMT (thiopurine methyltransferase) levels before starting to identify patients at risk for severe myelosuppression
- Mycophenolate mofetil (CellCept) — commonly used steroid-sparing agent; onset of action 3-6 months; generally well tolerated; teratogenic
- Tacrolimus — calcineurin inhibitor; faster onset than azathioprine (1-3 months); monitor renal function and drug levels
- Methotrexate — an alternative steroid-sparing agent; weekly dosing
- Cyclosporine — reserved for refractory cases; significant nephrotoxicity risk
- Cyclophosphamide — reserved for severe refractory MG; significant toxicity (bone marrow suppression, hemorrhagic cystitis, malignancy risk)
Targeted Biological Therapies
- Rituximab — anti-CD20 monoclonal antibody; depletes B cells; particularly effective in MuSK-MG, where it is considered a first-line disease-modifying therapy; also used in refractory AChR-MG
- Eculizumab (Soliris) — FDA-approved for generalized AChR-positive MG (2017); a complement C5 inhibitor that blocks formation of the membrane attack complex; administered intravenously every 2 weeks; highly effective but extremely expensive
- Ravulizumab (Ultomiris) — a longer-acting C5 inhibitor; FDA-approved for generalized AChR-positive MG; dosed every 8 weeks, reducing infusion burden
- Efgartigimod (Vyvgart) — FDA-approved in 2021; a neonatal Fc receptor (FcRn) inhibitor that accelerates IgG degradation, reducing pathogenic antibody levels; administered as IV infusion cycles; also available as subcutaneous formulation with hyaluronidase (Vyvgart Hytrulo)
- Rozanolixizumab — another FcRn inhibitor approved for generalized AChR-positive MG
- Zilucoplan — a subcutaneous complement C5 inhibitor; FDA-approved for generalized AChR-positive MG
Thymectomy
- Thymoma-associated MG — thymectomy is mandatory for all patients with thymoma regardless of MG severity; complete resection is essential
- Non-thymomatous AChR-positive generalized MG — the MGTX trial (2016) established that thymectomy (combined with prednisone) improves clinical outcomes, reduces prednisone requirements, and lowers hospitalization rates compared to prednisone alone; benefits persist for at least 5 years; most beneficial when performed early in the disease course, in patients aged 18-65, and within 5 years of onset
- Surgical approaches — transsternal extended thymectomy (traditional gold standard); minimally invasive approaches (video-assisted thoracoscopic surgery [VATS], robotic-assisted thymectomy) achieve comparable outcomes with less morbidity
- MuSK-MG — thymectomy is generally NOT recommended, as thymic pathology is typically absent
Management of Myasthenic Crisis
- ICU admission — mandatory; continuous monitoring of respiratory function (serial FVC, NIF measurements)
- Intubation and mechanical ventilation — when FVC falls below 15-20 mL/kg or clinical respiratory distress is evident; elective intubation is preferable to emergency intubation
- Intravenous immunoglobulin (IVIg) — 0.4 g/kg/day for 5 days; onset of improvement in 3-5 days; comparable efficacy to plasmapheresis
- Plasmapheresis (plasma exchange, PLEX) — 5 exchanges over 10-14 days; rapidly removes circulating antibodies; onset of improvement in 2-3 days; often preferred in severe crisis for its slightly faster onset
- Identify and treat precipitants — infections, offending medications, physiological stressors
- Avoid medications that worsen MG — aminoglycosides, fluoroquinolones, macrolides, beta-blockers, magnesium, phenytoin, procainamide, D-penicillamine, botulinum toxin, immune checkpoint inhibitors
8. Complications
- Myasthenic crisis — acute respiratory failure requiring mechanical ventilation; occurs in 15-20% of patients; mortality has decreased from >40% historically to approximately 3-5% with modern intensive care
- Cholinergic crisis — rare; caused by excessive acetylcholinesterase inhibitor dosing; presents with excessive secretions, miosis, fasciculations, and weakness; treatment involves withholding pyridostigmine and providing supportive care
- Aspiration pneumonia — from bulbar weakness and dysphagia
- Treatment-related complications — steroid side effects (osteoporosis, diabetes, infections, cataracts); immunosuppression-related infections; eculizumab-associated meningococcal infection risk
- Thymoma complications — local invasion, recurrence, associated paraneoplastic syndromes (pure red cell aplasia, hypogammaglobulinemia, Good syndrome)
- Neonatal myasthenia — transient weakness in newborns of MG mothers due to transplacental antibody transfer
- Arthrogryposis multiplex congenita — rare; caused by maternal antibodies inhibiting fetal AChR during development, leading to joint contractures
- Depression and anxiety — significantly increased prevalence; related to chronic disease burden and unpredictable symptom fluctuation
- Fatigue — pervasive complaint beyond muscular fatigability; impacts quality of life significantly
- Weight gain — from corticosteroid use and reduced physical activity
- Falls — from proximal and axial weakness
9. Prognosis
The prognosis of MG has improved dramatically over the past century. Before the advent of modern immunotherapy and intensive care, the mortality rate was approximately 30-40%. Today, with appropriate treatment, the mortality rate from MG itself is approximately 3-5%, and most patients achieve good symptom control with a near-normal life expectancy.
Treatment outcomes are classified using the MGFA Post-Intervention Status (PIS):
- Complete stable remission (CSR) — no symptoms, no treatment for at least 1 year; achieved by approximately 10-15% of patients
- Pharmacological remission — no symptoms on medication only; achieved by approximately 20-30%
- Minimal manifestation status (MMS) — no functional limitations but minor weakness detectable on examination; the practical treatment goal for most patients; achieved by approximately 40-60%
Factors associated with better prognosis include:
- Purely ocular disease — approximately 40-50% of patients with ocular MG remain ocular
- Younger age at onset
- Early thymectomy — particularly in young patients with AChR antibodies and thymic hyperplasia
- AChR antibody-positive subtype — generally more treatment-responsive than MuSK-MG
- Absence of thymoma
Factors associated with poorer prognosis include:
- Thymoma — associated with more refractory disease and risk of tumor recurrence
- MuSK-MG — historically more difficult to treat, though rituximab has significantly improved outcomes
- Late-onset disease with bulbar predominance
- Immune checkpoint inhibitor-triggered MG — often fulminant with high mortality (approximately 20-30%)
- Myasthenic crisis at presentation
10. Prevention
There are no established strategies to prevent the development of MG itself, as the underlying autoimmune trigger remains unknown. Prevention efforts focus on avoiding exacerbations, preventing myasthenic crisis, and minimizing treatment complications:
- Medication awareness — patients and physicians must be vigilant about medications that can worsen MG; all patients should carry a list of contraindicated drugs; medical alert identification is recommended
- Infection prevention — respiratory infections are the most common trigger for myasthenic crisis; patients should minimize exposure to sick contacts and practice good hand hygiene, particularly during respiratory virus season
- Stress management — emotional and physical stress can trigger exacerbations; patients benefit from stress reduction strategies and adequate rest
- Surgical precautions — before any surgery, anesthesiologists must be informed of the MG diagnosis; neuromuscular blocking agents should be used with extreme caution (reduced doses of non-depolarizing agents; resistance to succinylcholine)
- Pregnancy planning — MG can fluctuate during pregnancy; coordination between neurologist and obstetrician is essential; mycophenolate and methotrexate must be discontinued before conception
- Temperature management — heat can worsen symptoms; patients should avoid excessive heat exposure
- Osteoporosis prevention — calcium, vitamin D supplementation, and bone density monitoring for patients on long-term corticosteroids
Interactive Visualization The Neuromuscular Junction — fire a nerve at a muscle Fire a nerve and watch calcium dump acetylcholine across the gap to spark the muscle — then block it with Botox, strip the receptors in myasthenia gravis, or jam the off-switch with a nerve agent. Launch →
Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Research Papers
- Connections
- Featured Videos
Research Papers
Curated PubMed topic searches on Myasthenia Gravis. Each link opens a live PubMed query so the result set stays current as new studies are indexed.
- PubMed topic search: Myasthenia gravis review
- PubMed topic search: Acetylcholine receptor antibody myasthenia
- PubMed topic search: MuSK antibody myasthenia
- PubMed topic search: Thymectomy myasthenia gravis trial
- PubMed topic search: Pyridostigmine myasthenia
- PubMed topic search: Eculizumab myasthenia gravis
- PubMed topic search: Efgartigimod myasthenia trial
- PubMed topic search: Myasthenic crisis management
- PubMed topic search: Plasmapheresis IVIG myasthenia
- PubMed topic search: Ocular myasthenia gravis
- PubMed topic search: Thymoma myasthenia gravis
- PubMed topic search: Repetitive nerve stimulation myasthenia
Connections
- The Neuromuscular Junction: How a Nerve Commands a Muscle — interactive animation
- Magnesium
- Lupus
- Multiple Sclerosis
- ALS
- Peripheral Neuropathy
- Sjogren's Syndrome
- Vitamin B12
- Vitamin D3
- Alzheimer's Disease
- Parkinson's Disease
- Arthritis
- Fatigue
- Post-Viral Triggers: EBV and Others
- Stress Management
- Neuropsychiatric Lupus
- Biologics for Lupus
- Huntingtons Disease