Prion Disease
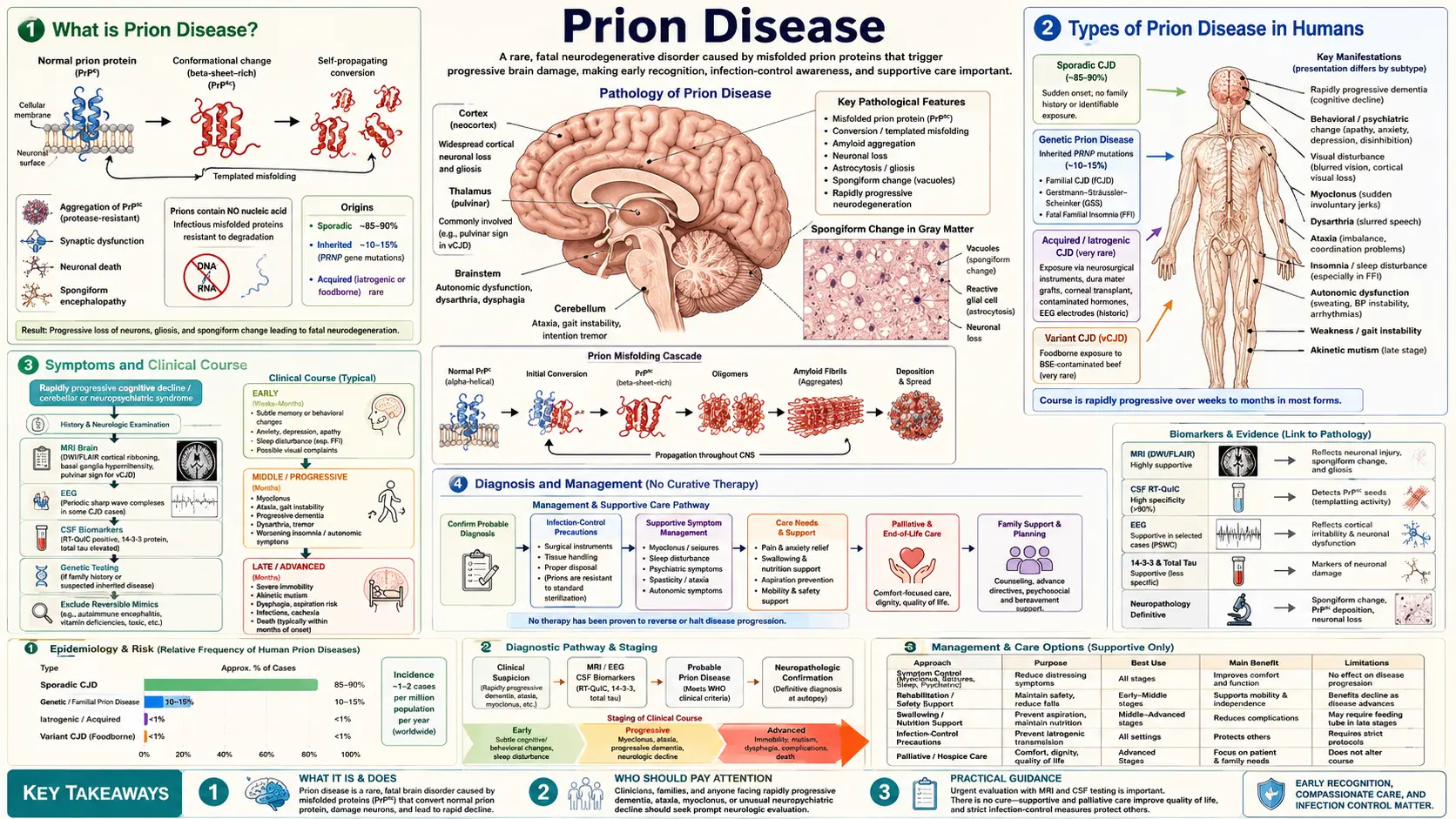
What is Prion Disease?
Prion disease is a group of rare, fatal, and rapidly progressive neurodegenerative disorders caused by misfolded prion proteins. The word "prion" is a portmanteau of proteinaceous infectious particle. Unlike bacteria, viruses, or fungi, prions contain no genetic material — they are purely proteins that propagate disease by inducing normal proteins to misfold and aggregate.
The cellular prion protein (PrPC) is a normal, membrane-anchored glycoprotein expressed throughout the body but most abundantly in the brain. In prion disease, PrPC converts to a misfolded, protease-resistant isoform called PrPSc (named after scrapie, the sheep form). Once present, PrPSc acts as a template that catalyzes the conversion of adjacent PrPC molecules in an exponential chain reaction, eventually destroying neurons and leaving characteristic spongiform (Swiss-cheese-like) vacuoles throughout the brain.
Types of Prion Disease in Humans
Human prion diseases are classified by their origin:
- Sporadic CJD (sCJD) — 85% of cases: No identified cause; arises from a spontaneous misfolding event. Incidence is approximately 1–2 per million people per year worldwide. Onset is typically age 50–75. Median survival is 4–6 months from symptom onset; 90% die within 12 months.
- Genetic (familial) CJD (gCJD) — ~15% of cases: Caused by autosomal dominant mutations in the PRNP gene on chromosome 20. Over 40 pathogenic mutations are known. Includes fatal familial insomnia (FFI), Gerstmann-Sträussler-Scheinker syndrome (GSS), and familial CJD. GSS typically presents in the 40s with cerebellar ataxia and a slower 2–10 year course.
- Iatrogenic CJD (iCJD) — <1% of cases: Transmitted through medical procedures: cadaveric pituitary-derived human growth hormone (now banned), dura mater grafts (particularly Lyodura brand), corneal transplants, contaminated neurosurgical instruments. Transmission from blood products has been documented only for variant CJD.
- Variant CJD (vCJD): Linked to consumption of bovine spongiform encephalopathy (BSE)-infected beef, primarily in the United Kingdom during the 1980s–1990s epidemic. Fewer than 250 confirmed cases worldwide. Affects younger patients (median age ~26 vs. ~65 for sCJD). Distinctive features: psychiatric and behavioral onset (months before neurological signs), painful sensory disturbances, and the "pulvinar sign" on MRI (bilateral posterior thalamic hyperintensity on FLAIR). Secondary transmission via blood transfusion from asymptomatic donors has occurred.
- Kuru: Historical form affecting the Fore people of Papua New Guinea through ritualistic cannibalism (now eradicated). Predominantly cerebellar presentation with a "trembling" course, 12–24 months to death. Studied by D. Carleton Gajdusek, who received the 1976 Nobel Prize.
Symptoms and Clinical Course
The hallmark of sporadic CJD is a rapidly progressive dementia (RPD) — cognitive decline measured in weeks to months rather than years. No other neurodegenerative disease deteriorates as fast.
Sporadic CJD cardinal features:
- Rapidly progressive dementia: Memory loss, confusion, disorientation that accelerates week over week.
- Cerebellar ataxia: Gait unsteadiness, loss of coordination, dysarthria.
- Myoclonus: Sudden, brief, shock-like muscle jerks — often triggered by a startling stimulus (startle myoclonus).
- Visual disturbances: Blurred vision, visual field defects, cortical blindness (Heidenhain variant).
- Pyramidal and extrapyramidal signs: Spasticity, hyperreflexia, rigidity.
- Akinetic mutism: Late-stage unresponsiveness — the patient is awake but motionless and mute.
Progression: prodromal phase of fatigue, anxiety, or sleep disturbance → rapid neurological decline over weeks → akinetic mutism → death. Median survival from symptom onset: 4–6 months (sCJD); up to 14 months (vCJD).
Diagnosis and Biomarkers
Prion disease is diagnosed by a combination of clinical presentation, EEG, MRI, and CSF biomarkers. Brain biopsy confirms the diagnosis definitively but is rarely performed in life.
- EEG — Periodic Sharp Wave Complexes (PSWCs): Biphasic or triphasic sharp waves occurring at approximately 1 Hz are found in ~67% of sporadic CJD cases. Sensitivity decreases with disease stage. PSWCs are not seen in vCJD.
- MRI — Diffusion-Weighted Imaging (DWI) / FLAIR: Cortical ribboning (gyral restricted diffusion along the cortex) and basal ganglia/thalamic hyperintensity on DWI are the most sensitive imaging findings, present in >90% of sCJD. The "pulvinar sign" (posterior thalamic hyperintensity) on FLAIR is specific for vCJD (>90% sensitivity in that subtype). MRI is now the most sensitive single test.
- CSF RT-QuIC (Real-Time Quaking-Induced Conversion): A highly sensitive amplification assay that detects seeding activity of misfolded prion protein in cerebrospinal fluid. Sensitivity 85–97%, specificity >99% for sCJD. This has largely replaced 14-3-3 protein as the preferred CSF test.
- CSF 14-3-3 Protein: A surrogate marker of rapid neuronal death. Sensitivity ~85% for sCJD; specificity lower (~90%) — can be positive in any cause of acute, massive neuronal loss (encephalitis, ischemic stroke). Still used but less specific than RT-QuIC.
- CSF tau and neurogranin: Markedly elevated total tau (>1,300 pg/mL) is a strong supportive finding for CJD. Phospho-tau is typically normal or only mildly elevated, distinguishing CJD from Alzheimer's disease.
- PRNP genetic testing: All suspected cases should have PRNP sequencing to identify mutations (gCJD) and codon 129 polymorphism (MM/MV/VV) — codon 129 MM homozygotes are more susceptible to both sCJD and vCJD.
- Nasal brushings RT-QuIC: A non-invasive alternative using olfactory mucosa; sensitivity ~97% in confirmed sCJD — may eventually replace lumbar puncture as initial testing.
- Prion surveillance: All suspected prion disease cases are reportable to public health authorities in the United States (CDC National Prion Disease Pathology Surveillance Center) and most other countries. Brain autopsy is strongly encouraged to confirm diagnosis and subtype.
Molecular Mechanism of Prion Propagation
The prion protein gene PRNP encodes a 253-amino-acid precursor that is post-translationally modified into the mature GPI-anchored PrPC. PrPC has a predominantly alpha-helical secondary structure and is normally expressed on neurons, astrocytes, and many peripheral tissues. Its physiological function remains incompletely understood but involves copper binding, circadian rhythm regulation, and neuroprotection.
PrPSc, the disease isoform, has a high proportion of beta-sheet content, rendering it partially resistant to proteinase K digestion and detergent solubilization. When a molecule of PrPSc contacts PrPC, it catalyzes a conformational change — the core mechanism of prion propagation. This process occurs exponentially: one seed generates two, two generate four, and so on, until the neuronal burden becomes lethal.
Different prion "strains" exist despite identical primary sequences. Strain identity is encoded in the three-dimensional conformation of PrPSc — a phenomenon with no parallel in classical infectious disease. Strains differ in incubation period, regional brain tropism, and proteinase-K cleavage pattern (termed "prion type" 1 or 2 in sCJD). Type 2 PrPSc combined with codon 129 MV genotype defines the most common sCJD subtype (MM1/MV1).
Decontamination and Infection Control
PrPSc is extraordinarily resistant to standard sterilization methods that inactivate all conventional pathogens:
- Ineffective: Routine autoclaving (121°C), formaldehyde fixation, ethanol, glutaraldehyde, bleach at standard concentrations, ultraviolet radiation, gamma irradiation, boiling, dry heat up to 160°C, and nuclease treatment (there is no nucleic acid to destroy).
- Effective decontamination:
- Sodium hydroxide (NaOH) 1 M (1–2 hours) + gravity autoclave (134°C, 1 hour): The WHO-endorsed standard for prion decontamination of reusable surgical instruments. NaOH alone or autoclaving alone is insufficient.
- Sodium hypochlorite 20,000 ppm (2% bleach) for 1 hour: Acceptable chemical-only alternative for heat-sensitive materials.
- Single-use instruments: For procedures on high-risk brain/posterior eye/spinal cord tissue in patients with suspected prion disease.
- Autopsy precautions: Full PPE (face shield, double gloves, impermeable gown), dedicated instruments (autoclaved or incinerated), formalin fixation followed by formic acid treatment before routine processing.
- Blood/organ donation: Patients with known or suspected prion disease are permanently deferred from blood, tissue, and organ donation. vCJD presents specific blood-supply challenges because of asymptomatic carrier transmission.
Treatment and Prognosis
There is currently no proven disease-modifying treatment for any human prion disease. All confirmed cases are uniformly fatal. Management is palliative:
- Symptomatic relief: Clonazepam or valproate for myoclonus; antipsychotics for agitation in vCJD; opioids for pain; artificial nutrition and hydration per patient/family wishes.
- Supportive care: Aspiration precautions, skin care, bowel and bladder management, caregiver support and hospice planning.
- Investigational approaches: Quinacrine and pentosan polysulfate showed no benefit in clinical trials. Antisense oligonucleotides (ASOs) targeting PRNP mRNA have demonstrated efficacy in animal models and entered Phase I trials. Immunotherapy targeting PrP is also in early development. The challenge is the extremely rapid progression — the therapeutic window is narrow.
- Genetic counseling: Families with gCJD should receive genetic counseling. Presymptomatic testing raises complex ethical questions; a positive PRNP mutation test is essentially a sentence of future death from an untreatable disease.
Prognosis by subtype: sCJD median survival 4–6 months; vCJD median 13–14 months; GSS 2–10 years; FFI 7–36 months; kuru historically 12–24 months.
Animal Prion Diseases and Zoonotic Risk
Prion diseases affect many animal species:
- Scrapie (sheep and goats): The first recognized prion disease, described in 1732. Scrapie does not appear to cause disease in humans despite centuries of exposure.
- Bovine Spongiform Encephalopathy (BSE, "mad cow disease"): UK epidemic 1980s–1990s, caused by feeding cattle rendered protein from scrapie-infected sheep. BSE prions crossed the species barrier to humans as variant CJD.
- Chronic Wasting Disease (CWD): Affects cervids (deer, elk, moose, caribou) and is spreading across North America, Europe, and Asia. CWD prions are highly transmissible among cervids via saliva, urine, and feces. To date, no confirmed human cases exist, but primate studies suggest the species barrier is not absolute. The CDC recommends against eating meat from CWD-positive animals. CWD represents a significant ongoing public health concern.
- Feline Spongiform Encephalopathy (FSE): Cats exposed to BSE-contaminated pet food during the UK epidemic.
Risk Factors and Prevention
- Codon 129 genotype: MM homozygosity at codon 129 of PRNP is over-represented in sCJD and vCJD patients (80–90% of cases vs. ~37% in the general population). VV homozygosity and MV heterozygosity confer relative protection — heterozygosity is thought to have been under positive selection in populations with historical cannibalism (Fore people show high rates of protective polymorphisms).
- PRNP mutations (gCJD): Family members of gCJD patients should consider genetic testing and counseling.
- Medical exposure (iCJD): Eliminated through: ban on cadaveric pituitary growth hormone (1985 in US), discontinuation of implicated dura mater products, sterilization protocol upgrades, and surgical instrument tracking for high-risk procedures.
- Dietary precautions: Avoiding brain and spinal cord from cattle in BSE-affected regions. CWD: do not hunt or consume cervids from CWD-endemic areas; test harvested animals before consuming.
- No person-to-person transmission by casual contact, respiratory route, or fecal-oral route has ever been documented. Standard infection control (gloves, hand washing) is sufficient for routine patient care.
Table of Contents
- What is Prion Disease?
- Types of Prion Disease in Humans
- Symptoms and Clinical Course
- Diagnosis and Biomarkers
- Molecular Mechanism
- Decontamination and Infection Control
- Treatment and Prognosis
- Animal Prion Diseases and Zoonotic Risk
- Risk Factors and Prevention
- Research Papers
- Connections
- Featured Videos
Research Papers
Curated PubMed citations and topic searches on prion disease. PMID links open the specific article; topic-search links stay current as new studies are indexed.
- Prusiner SB. Prions. PNAS. 1998. PMID 14522853
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- PubMed topic search: PRNP antisense oligonucleotide prion therapy
Connections
- Neurology
- Alzheimer's Disease
- Parkinson's Disease
- Huntington's Disease
- ALS
- Lewy Body Dementia
- Frontotemporal Dementia
- Multiple Sclerosis
- Peripheral Neuropathy
- Vitamin B12
- CSF Analysis
- Depression
- Restless Legs Syndrome