Huntington's Disease
1. Overview
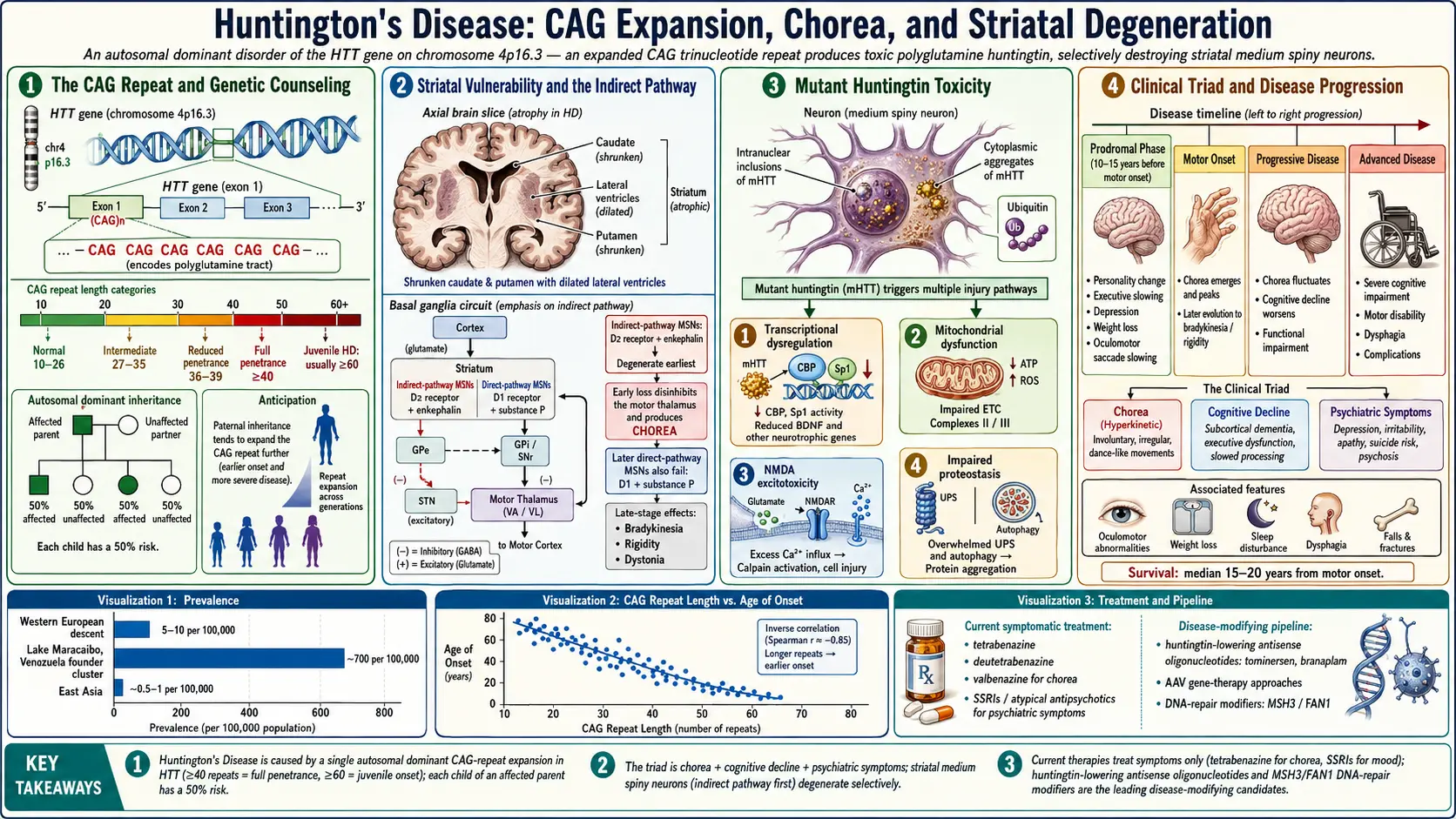
Huntington's disease (HD) is a progressive, fatal neurodegenerative disorder caused by an autosomal dominant mutation in the huntingtin (HTT) gene on chromosome 4p16.3. The mutation consists of an expanded CAG trinucleotide repeat that encodes an abnormally long polyglutamine (polyQ) tract in the huntingtin protein, leading to protein misfolding, aggregation, and selective neuronal death, particularly in the striatum (caudate nucleus and putamen) and cerebral cortex.
The disease is characterized by a classic triad of motor dysfunction (chorea and other movement abnormalities), cognitive decline (progressing to dementia), and psychiatric disturbances (depression, irritability, apathy, and psychosis). Symptoms typically emerge between ages 30 and 50, though the range extends from childhood to old age. Once symptoms begin, the disease progresses relentlessly over 15-20 years, ultimately leading to complete disability and death.
Huntington's disease is classified based on the number of CAG repeats and the clinical stage:
- Normal — 10-26 CAG repeats; no disease risk
- Intermediate (mutable normal) — 27-35 repeats; individual is unaffected but expansion may occur during transmission to offspring
- Reduced penetrance — 36-39 repeats; some individuals develop the disease, others do not
- Full penetrance — 40 or more repeats; disease will develop if the individual lives long enough
- Juvenile Huntington's disease (JHD) — onset before age 20; typically associated with very large repeat expansions (usually 60 or more); accounts for approximately 5-10% of cases
2. Epidemiology
Huntington's disease affects approximately 5-10 per 100,000 people in populations of Western European descent, making it one of the most common autosomal dominant neurodegenerative disorders. The prevalence is highest in populations of European ancestry, particularly in the Lake Maracaibo region of Venezuela, where the prevalence exceeds 700 per 100,000 due to a founder effect traced to a single affected individual in the early 19th century. In contrast, the prevalence in Japan, China, and sub-Saharan Africa is significantly lower, approximately 0.5-1 per 100,000.
In the United States, an estimated 30,000 individuals have symptomatic HD, with an additional 200,000 at risk of having inherited the gene. There is no sex predilection, as the autosomal dominant inheritance pattern affects males and females equally. However, paternal transmission is associated with a higher risk of anticipation (expansion of the CAG repeat during spermatogenesis), leading to earlier onset in the next generation. This is particularly pronounced in juvenile HD, where more than 80% of cases are paternally inherited.
The mean age of onset is approximately 40 years, though it can range from 2 to over 80 years. There is a strong inverse correlation between CAG repeat length and age of onset, with repeat length accounting for approximately 50-70% of the variance in onset age. The remaining variance is influenced by genetic modifiers, environmental factors, and stochastic events.
3. Pathophysiology
The pathophysiology of Huntington's disease centers on the toxic effects of the mutant huntingtin protein (mHTT) containing an expanded polyglutamine tract. The huntingtin protein is ubiquitously expressed, but the striatum and cortex are selectively vulnerable to its toxic effects.
Mutant Huntingtin Protein Toxicity
The expanded polyglutamine tract causes the huntingtin protein to adopt abnormal conformations, leading to the formation of intranuclear inclusions and cytoplasmic aggregates. These inclusions contain mHTT, ubiquitin, and components of the proteasomal and autophagy degradation machinery. The relationship between visible aggregates and toxicity is complex; soluble oligomeric and misfolded mHTT species are now believed to be more toxic than the large insoluble inclusions, which may represent a protective sequestration mechanism. mHTT disrupts multiple cellular processes including gene transcription, protein homeostasis, mitochondrial function, axonal transport, and synaptic signaling.
Striatal Vulnerability and the Indirect Pathway
The medium spiny neurons (MSNs) of the striatum are the most vulnerable cell population in HD. MSNs expressing dopamine D2 receptors and enkephalin (indirect pathway) degenerate earlier than those expressing D1 receptors and substance P (direct pathway). Early loss of indirect pathway neurons disinhibits the motor thalamus, leading to chorea (excessive, involuntary, dance-like movements). As the disease progresses and both pathways degenerate, chorea gives way to bradykinesia, rigidity, and dystonia. Large cholinergic interneurons and NADPH-diaphorase-positive interneurons are relatively spared.
Transcriptional Dysregulation
mHTT interacts with and sequesters key transcription factors, including CREB-binding protein (CBP), Sp1, TAFII130, and the REST/NRSF silencing complex. Normal huntingtin sequesters REST/NRSF in the cytoplasm, allowing neuronal gene expression. mHTT fails to bind REST/NRSF, allowing it to translocate to the nucleus and repress neuronal genes, including brain-derived neurotrophic factor (BDNF). Reduced BDNF, which is produced primarily in the cortex and transported to the striatum, contributes significantly to MSN vulnerability.
Mitochondrial Dysfunction and Excitotoxicity
mHTT directly impairs mitochondrial function by associating with the outer mitochondrial membrane, disrupting the electron transport chain (particularly complexes II and III), reducing ATP production, and increasing oxidative stress. The resulting energy deficit renders MSNs more susceptible to glutamate-mediated excitotoxicity via NMDA receptors, particularly those containing the NR2B subunit. This excitotoxic hypothesis is supported by the observation that intrastriatal injection of the mitochondrial toxin 3-nitropropionic acid replicates many features of HD pathology.
Impaired Proteostasis
mHTT overwhelms the cell's protein quality control systems, including the ubiquitin-proteasome system (UPS) and autophagy pathways. The proteasome cannot efficiently degrade the expanded polyglutamine tract, and mHTT aggregates physically impair proteasome function. Autophagy, while upregulated as a compensatory mechanism, becomes inefficient as mHTT impairs cargo recognition by autophagosomes. This proteostatic failure contributes to the accumulation of toxic protein species and cellular dysfunction.
Cortical Pathology
While striatal atrophy is the hallmark of HD, the cerebral cortex undergoes significant degeneration, particularly in layers III, V, and VI. Cortical thinning, especially in the prefrontal, premotor, and sensorimotor regions, correlates with cognitive and motor symptom severity. The loss of corticostriatal projections contributes to striatal dysfunction through both loss of trophic support (BDNF) and altered glutamatergic input.
4. Etiology and Risk Factors
Genetic Cause
Huntington's disease is caused exclusively by an expanded CAG trinucleotide repeat in exon 1 of the HTT gene on chromosome 4p16.3. The inheritance is autosomal dominant with age-dependent penetrance. Each child of an affected parent has a 50% chance of inheriting the expanded allele.
- CAG repeat length is the primary determinant of disease, with longer repeats associated with earlier onset
- Anticipation — the tendency for CAG repeat length to increase during transmission, particularly through paternal inheritance (due to instability during spermatogenesis)
- De novo mutations — approximately 5-10% of HD cases have no family history and arise from expansion of intermediate alleles (27-35 repeats) into the pathogenic range
- Somatic mosaicism — the CAG repeat length can differ between tissues within the same individual; the brain, particularly the striatum, tends to show longer repeats than blood, contributing to selective vulnerability
Genetic Modifiers of Onset Age
While CAG repeat length is the strongest predictor of onset age, genetic modifier studies have identified several DNA repair pathway genes that influence the age of clinical manifestation:
- FAN1 (FANCD2 and FANCI Associated Nuclease 1) — variants accelerate or delay onset
- MSH3 (MutS Homolog 3) — a mismatch repair gene; its activity promotes somatic expansion of the CAG repeat
- PMS1, PMS2, MLH1 — additional mismatch repair genes implicated as modifiers
- LIG1 (DNA Ligase 1) — involved in DNA repair
Environmental Modifiers
- Physical exercise — associated with later onset and slower progression in some observational studies
- Cognitive reserve — higher education and cognitive engagement may delay symptom onset
- Head trauma — may accelerate onset in gene carriers (limited evidence)
- Stress and lifestyle factors — their role as modifiers remains under investigation
5. Clinical Presentation
Prodromal Phase
Before the formal diagnosis of motor onset, gene carriers may experience a prodromal phase lasting 10-15 years, during which subtle changes in cognition, mood, and motor function are detectable on sensitive testing. These include:
- Subtle personality changes and irritability
- Mild cognitive slowing, particularly in executive functions
- Depression and anxiety
- Subtle motor abnormalities on quantitative testing
- Weight loss beginning years before motor onset
Motor Symptoms
- Chorea — the hallmark motor feature; involuntary, irregular, flowing, dance-like movements affecting the face, trunk, and limbs; typically begins subtly and worsens over 10-15 years before declining as rigidity predominates
- Dystonia — sustained abnormal postures, particularly in advanced disease and juvenile HD
- Bradykinesia — progressive slowing of voluntary movement, increasingly prominent in later stages
- Rigidity — increased muscle tone, predominant in the Westphal (juvenile akinetic-rigid) variant
- Gait impairment — wide-based, unsteady gait with increased fall risk; a leading cause of injury
- Oculomotor dysfunction — impaired saccadic eye movements (slowed initiation and velocity) are among the earliest motor signs
- Dysarthria — progressive impairment of speech clarity, rate, and volume
- Dysphagia — difficulty swallowing, increasing aspiration risk in advanced disease
- Motor impersistence — inability to sustain a voluntary motor act, such as tongue protrusion or grip
Cognitive Symptoms
- Executive dysfunction — the earliest and most prominent cognitive domain affected; impaired planning, cognitive flexibility, judgment, and abstract thinking
- Psychomotor slowing — reduced speed of information processing
- Visuospatial deficits — impaired spatial perception and mental rotation
- Memory impairment — primarily retrieval deficits (recognition memory relatively preserved compared to Alzheimer's disease); procedural memory is severely affected
- Reduced awareness (anosognosia) — many patients underestimate the severity of their symptoms
- Language — word-finding difficulties develop later; comprehension is relatively preserved until advanced stages
- Dementia — progressive subcortical dementia develops in virtually all patients
Psychiatric Symptoms
- Depression — affects approximately 40-50% of patients; can precede motor onset by years; significantly contributes to functional disability
- Irritability and aggression — among the most challenging symptoms for caregivers; affects 40-70% of patients
- Apathy — progressive loss of motivation and initiative; distinct from depression; affects 50-70% of patients
- Anxiety — affects approximately 30-40% of patients
- Obsessive-compulsive behaviors — rigidity of thought, perseveration, and repetitive behaviors
- Psychosis — hallucinations and delusions in approximately 5-10% of patients
- Suicidality — the suicide rate in HD is 4-6 times that of the general population; highest risk at the time of genetic testing disclosure and at onset of functional decline
Juvenile Huntington's Disease (Westphal Variant)
Juvenile HD (onset before age 20) presents differently from adult-onset HD:
- Rigidity and bradykinesia dominate rather than chorea (akinetic-rigid or "Westphal" variant)
- Seizures — occur in 30-50% of juvenile cases (rare in adult-onset HD)
- Rapid cognitive decline — school performance deterioration is often the presenting feature
- Cerebellar ataxia — more prominent than in adult-onset disease
- More rapid progression — mean disease duration approximately 10 years
6. Diagnosis
Clinical Diagnosis
The diagnosis of manifest (motor-onset) HD requires:
- The presence of unequivocal motor signs (typically chorea, but also dystonia, bradykinesia, or oculomotor dysfunction) in an individual with a known family history of HD or a positive genetic test
- Motor onset is formally defined by a score of 4 (motor abnormalities that are unequivocal signs of HD) on the Unified Huntington's Disease Rating Scale (UHDRS) Diagnostic Confidence Level
Genetic Testing
- Confirmatory genetic testing — PCR-based assay to determine the number of CAG repeats in the HTT gene; the gold standard for diagnosis
- Presymptomatic (predictive) testing — available for at-risk individuals who wish to know their genetic status; conducted under strict guidelines established by the International Huntington Association (IHA) and the World Federation of Neurology (WFN), including mandatory genetic counseling, psychological assessment, and a minimum reflection period
- Prenatal testing — available through chorionic villus sampling or amniocentesis
- Preimplantation genetic diagnosis (PGD) — allows selection of unaffected embryos during in vitro fertilization; can be performed with or without revealing the at-risk parent's gene status (exclusion testing)
Neuroimaging
- MRI — shows progressive caudate nucleus atrophy with enlargement of the frontal horns of the lateral ventricles ("box-car" ventricles); caudate atrophy can be detected years before motor onset in premanifest gene carriers; also reveals cortical thinning and white matter changes
- Volumetric MRI — quantitative measurement of striatal and cortical volume is used as a biomarker in clinical trials
- PET and SPECT — reduced striatal glucose metabolism (FDG-PET) and reduced dopamine D2 receptor binding (raclopride-PET) can be detected years before clinical onset
- fMRI — altered activation patterns in corticostriatal circuits during cognitive tasks
Clinical Assessment Tools
- Unified Huntington's Disease Rating Scale (UHDRS) — the standard clinical assessment tool; includes motor, cognitive (Symbol Digit Modalities Test, Stroop tests, verbal fluency), behavioral, and functional capacity subscales
- Total Functional Capacity (TFC) scale — rates independence in occupation, finances, domestic chores, activities of daily living, and care level; scores range from 13 (normal) to 0 (total dependence); used for clinical staging:
- Stage I: TFC 11-13
- Stage II: TFC 7-10
- Stage III: TFC 3-6
- Stage IV: TFC 1-2
- Stage V: TFC 0
- Problem Behaviors Assessment (PBA) — structured assessment of psychiatric symptoms specific to HD
- HD-CAB (Huntington's Disease Cognitive Assessment Battery) — standardized cognitive battery for HD clinical trials
Biomarkers
- Neurofilament light chain (NfL) — elevated in blood and CSF; correlates with disease stage, brain atrophy, and rate of progression; the most promising fluid biomarker for HD
- Mutant huntingtin protein (mHTT) — detectable in CSF using ultrasensitive immunoassays; correlates with disease progression
7. Treatment
There is currently no disease-modifying therapy for Huntington's disease. Treatment is entirely symptomatic and requires a multidisciplinary team approach involving neurology, psychiatry, genetics, rehabilitation, speech therapy, nutrition, and social work.
Treatment of Chorea
- Tetrabenazine (Xenazine) — the first FDA-approved drug for HD chorea (2008); a vesicular monoamine transporter 2 (VMAT2) inhibitor that depletes presynaptic dopamine; dose titrated up to 75-100 mg/day; side effects include depression, parkinsonism, sedation, and akathisia
- Deutetrabenazine (Austedo) — FDA-approved in 2017; a deuterated form of tetrabenazine with longer half-life, allowing twice-daily dosing and improved tolerability; dose up to 48 mg/day
- Valbenazine (Ingrezza) — another VMAT2 inhibitor used off-label
- Antipsychotics — olanzapine, risperidone, haloperidol; may also address irritability and psychosis; used when chorea is accompanied by psychiatric symptoms
- Benzodiazepines — clonazepam for mild chorea and anxiety; sedation limits use
- Amantadine — NMDA antagonist with modest anti-choreic effects; limited evidence
Treatment of Psychiatric Symptoms
- Depression — SSRIs (citalopram, sertraline, fluoxetine) are first-line; venlafaxine and mirtazapine are alternatives; electroconvulsive therapy (ECT) for severe refractory depression
- Irritability and aggression — SSRIs, atypical antipsychotics (olanzapine, quetiapine), mood stabilizers (valproic acid, carbamazepine)
- Apathy — the most difficult symptom to treat; bupropion and psychostimulants may provide modest benefit; environmental structuring and scheduled activities
- Anxiety — SSRIs, buspirone, benzodiazepines (short-term use)
- Psychosis — atypical antipsychotics (olanzapine, risperidone, quetiapine); typical antipsychotics may worsen motor symptoms
- Obsessive-compulsive behaviors — SSRIs at higher doses, cognitive behavioral therapy
- Insomnia — melatonin, trazodone, mirtazapine; sleep hygiene education
Treatment of Other Motor Symptoms
- Dystonia — botulinum toxin injections for focal dystonia; benzodiazepines; baclofen
- Myoclonus — clonazepam, valproic acid, levetiracetam
- Rigidity and bradykinesia (late-stage) — levodopa or dopamine agonists may be tried but are often ineffective; amantadine
- Seizures (juvenile HD) — valproic acid, levetiracetam, lamotrigine
Supportive Care
- Physical therapy — balance training, gait exercises, stretching, fall prevention strategies; exercise programs have shown benefits for motor function and mood
- Speech therapy — strategies for dysarthria and dysphagia; communication aids in advanced disease
- Occupational therapy — home safety modifications, adaptive equipment, strategies for activities of daily living
- Nutritional management — high-calorie diet to counteract the hypermetabolic state and weight loss characteristic of HD; modified diet textures for dysphagia; percutaneous gastrostomy (PEG) when oral intake becomes unsafe
- Palliative care — advance care planning, end-of-life discussions, hospice referral
- Genetic counseling — for patients and family members; support for presymptomatic testing decisions
- Psychosocial support — patient and caregiver support groups; respite care; long-term care planning
8. Complications
- Aspiration pneumonia — the leading cause of death; results from progressive dysphagia and impaired cough reflex
- Falls and injuries — due to chorea, gait instability, and impaired judgment; subdural hematoma is a significant risk
- Severe malnutrition and cachexia — from dysphagia, hypermetabolism, and the catabolic state of HD
- Suicide — significantly elevated risk; requires ongoing psychiatric monitoring and safety assessment
- Cardiovascular disease — emerging evidence suggests increased cardiac mortality, possibly related to mutant huntingtin expression in cardiac tissue
- Pressure ulcers — in immobile, advanced-stage patients
- Contractures — from progressive rigidity and immobility in advanced disease
- Venous thromboembolism — from immobility
- Social isolation and family disruption — the combined effects of cognitive, behavioral, and motor impairment profoundly impact family dynamics
- Caregiver burnout — HD caregiving is exceptionally demanding due to the long disease duration and behavioral symptoms
- Dental disease — from impaired oral hygiene, dyskinesias, and medication side effects
9. Prognosis
Huntington's disease is invariably fatal. The median survival from motor onset to death is 15-20 years, with a range of 5-30 years. The mean age at death is approximately 55-60 years. The leading causes of death are aspiration pneumonia (approximately 33-40%), cardiovascular disease (approximately 25%), and suicide (approximately 7-10%).
Factors associated with more rapid progression include:
- Longer CAG repeat length — particularly repeats above 50
- Younger age at onset — juvenile HD has a more aggressive course with mean duration of approximately 10 years
- Predominantly rigid phenotype — associated with faster functional decline than chorea-predominant presentation
- Greater weight loss — an independent predictor of faster decline
- Higher baseline neurofilament light chain levels
Functional decline follows a relatively predictable trajectory. Patients typically become unable to work within 5-8 years of motor onset, require assistance with activities of daily living by 8-12 years, and become fully dependent by 12-18 years. The Total Functional Capacity (TFC) score declines by approximately 0.5-1.0 points per year.
10. Prevention
Because Huntington's disease is caused by a single fully penetrant genetic mutation, primary prevention focuses on reproductive options and genetic counseling:
- Genetic counseling — comprehensive counseling for at-risk individuals and families, covering inheritance patterns, testing options, implications, and reproductive choices
- Presymptomatic genetic testing — allows at-risk individuals to learn their gene status; must be performed with proper counseling and psychological support per international guidelines; approximately 5-20% of at-risk individuals choose to undergo predictive testing
- Preimplantation genetic diagnosis (PGD) — during in vitro fertilization, embryos are tested and only those without the expanded CAG repeat are implanted; prevents transmission to the next generation
- Prenatal diagnosis — chorionic villus sampling or amniocentesis can determine the fetal genotype; raises difficult decisions about pregnancy continuation
- Exclusion testing — for grandchildren of affected individuals who do not wish to know the at-risk parent's status; uses linkage analysis to determine whether the fetus inherited the chromosome from the affected or unaffected grandparent
For individuals who carry the gene, potential disease-delaying strategies under investigation include:
- Regular physical exercise — observational studies suggest association with later onset and slower progression
- Cognitive engagement — maintaining cognitive reserve through education and intellectually stimulating activities
- Mediterranean or anti-inflammatory diet — under investigation; no definitive evidence yet
- Avoidance of excessive alcohol and drug use — which may accelerate neurodegeneration
Interactive Visualization Protein Folding — fold it right, or watch it clump Watch a chaperone help a new protein fold into shape — then take it away and see it clump into the aggregates behind Alzheimer's and Huntington's, or a prion convert its neighbours. Launch →
Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Research Papers
- Connections
- Featured Videos
Research Papers
Curated PubMed topic searches on Huntington's Disease. Each link opens a live PubMed query so the result set stays current as new studies are indexed.
- PubMed topic search: Huntington disease review
- PubMed topic search: CAG repeat expansion Huntington
- PubMed topic search: Huntingtin protein aggregation
- PubMed topic search: Huntington disease predictive testing
- PubMed topic search: Tetrabenazine chorea Huntington
- PubMed topic search: Deutetrabenazine Huntington trial
- PubMed topic search: Tominersen Huntington antisense
- PubMed topic search: Huntington disease neuropathology striatum
- PubMed topic search: Juvenile Huntington disease
- PubMed topic search: Huntington disease biomarkers
- PubMed topic search: UHDRS rating scale Huntington
- PubMed topic search: Huntington disease cognitive decline
Connections
- How Proteins Fold — and Misfold in Disease — interactive animation
- Alzheimer's Disease
- Parkinson's Disease
- ALS
- Dementia
- Depression
- Anxiety
- Lion's Mane Mushroom
- Magnesium
- Vitamin B12
- Multiple Sclerosis
- Myasthenia Gravis
- Peripheral Neuropathy
- Sleep Hygiene
- Anti-Inflammatory Diet
- Oxidative Stress