Amyotrophic Lateral Sclerosis (ALS)
1. Overview
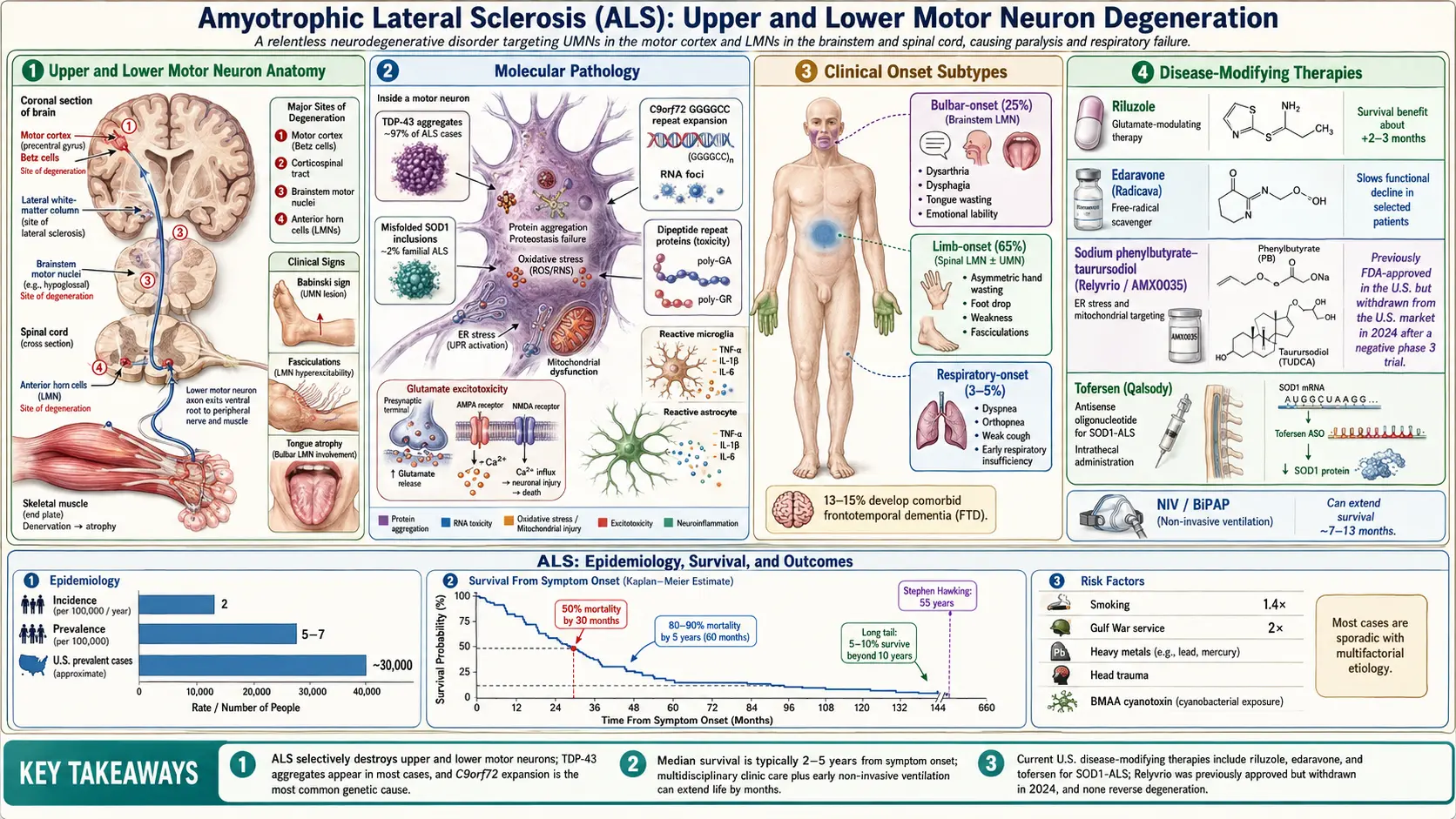
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease or motor neuron disease (MND), is a progressive neurodegenerative disorder that selectively attacks upper motor neurons (UMNs) in the motor cortex and lower motor neurons (LMNs) in the brainstem and spinal cord. The relentless degeneration of these neurons leads to progressive muscle weakness, atrophy, fasciculations, and spasticity, ultimately resulting in paralysis and death, most commonly from respiratory failure. ALS is the most common form of motor neuron disease in adults.
The disease was first described by French neurologist Jean-Martin Charcot in 1869, but it gained widespread public awareness in the United States after it struck the legendary baseball player Lou Gehrig in 1939. The name "amyotrophic" refers to muscle atrophy and weakness ("a-" without, "myo-" muscle, "trophic" nourishment), while "lateral sclerosis" describes the hardening of the lateral columns of the spinal cord where the corticospinal tracts degenerate.
ALS is classified into several subtypes based on clinical and genetic features:
- Sporadic ALS (sALS) — accounts for approximately 90-95% of cases with no clear family history
- Familial ALS (fALS) — accounts for 5-10% of cases with an identifiable genetic mutation inherited in an autosomal dominant pattern
- Limb-onset ALS — begins with weakness in the arms or legs (approximately 65% of cases)
- Bulbar-onset ALS — begins with speech or swallowing difficulties (approximately 25% of cases)
- Respiratory-onset ALS — rare presentation beginning with respiratory insufficiency (approximately 3-5% of cases)
- ALS-FTD — ALS co-occurring with frontotemporal dementia, seen in approximately 15% of patients
2. Epidemiology
ALS affects approximately 2 per 100,000 people per year worldwide, with a prevalence of 5-7 per 100,000. In the United States, an estimated 30,000 individuals are living with ALS at any given time, with roughly 5,000 new cases diagnosed annually. The disease is approximately 1.3-1.5 times more common in men than in women, although this sex difference diminishes with advancing age.
The peak age of onset for sporadic ALS is between 55 and 75 years, with a mean age of onset of approximately 63 years. Familial ALS tends to present earlier, with a mean onset around 46 years. ALS is rare before age 25, though juvenile-onset cases do occur. The incidence appears relatively uniform across European and North American populations but is notably higher in certain geographic clusters, including the Kii Peninsula of Japan, western New Guinea, and Guam, where it is associated with the ALS-parkinsonism-dementia complex (ALS-PDC).
The lifetime risk of developing ALS is estimated at approximately 1 in 300 to 1 in 400. The incidence in Europe and North America has been relatively stable over the past several decades, though some studies suggest a modest increase, likely attributable to improved diagnostic ascertainment and aging populations. Military veterans, particularly those who served in the Gulf War, have been reported to have a nearly two-fold increased risk of developing ALS.
3. Pathophysiology
The pathophysiology of ALS is multifactorial and involves a complex interplay of molecular and cellular mechanisms that converge to produce motor neuron degeneration. The disease selectively targets motor neurons while relatively sparing sensory neurons, autonomic neurons, and oculomotor neurons.
Protein Misfolding and Aggregation
A hallmark of ALS pathology is the accumulation of misfolded protein aggregates within motor neurons. The most common inclusion is TDP-43 (TAR DNA-binding protein 43), found in approximately 97% of all ALS cases. TDP-43 is normally a nuclear RNA-binding protein, but in ALS it becomes hyperphosphorylated, ubiquitinated, and mislocalized to the cytoplasm, where it forms insoluble aggregates. In cases caused by SOD1 mutations, misfolded SOD1 protein aggregates are the primary inclusion. FUS (fused in sarcoma) protein aggregates are found in a smaller subset of cases.
Glutamate Excitotoxicity
Motor neurons in ALS are subjected to excessive glutamate signaling, leading to sustained calcium influx through AMPA and NMDA receptors. This excitotoxicity is partly due to reduced expression and function of the excitatory amino acid transporter 2 (EAAT2/GLT-1) on astrocytes, which normally clears synaptic glutamate. The resulting intracellular calcium overload activates destructive enzymatic cascades including calpains, endonucleases, and phospholipases. This mechanism forms the basis for the therapeutic action of riluzole.
Oxidative Stress
Motor neurons exhibit heightened vulnerability to oxidative damage due to their high metabolic demands, large cell body size, and long axonal projections. Mutations in SOD1 (superoxide dismutase 1) lead to a toxic gain-of-function that generates reactive oxygen species (ROS), causing lipid peroxidation, protein oxidation, and DNA damage. Mitochondrial dysfunction further amplifies oxidative stress through impaired electron transport chain activity and increased superoxide production.
Neuroinflammation
Activated microglia and reactive astrocytes play a dual role in ALS pathogenesis. Initially, microglia may be neuroprotective (M2 phenotype), but as the disease progresses, they shift to a neurotoxic (M1 phenotype) state, releasing pro-inflammatory cytokines including TNF-alpha, IL-1beta, and IL-6. Reactive astrocytes lose their supportive functions and acquire toxic properties, actively contributing to motor neuron death through mechanisms involving NF-kB signaling.
Impaired Axonal Transport and Cytoskeletal Dysfunction
Motor neurons depend on efficient axonal transport to shuttle organelles, proteins, and trophic factors along axons that can extend over one meter in length. ALS disrupts both anterograde and retrograde axonal transport through damage to neurofilaments, dynein-dynactin complexes, and kinesin motors. Accumulation of neurofilaments in proximal axons is an early pathological finding and contributes to axonal dysfunction and eventual degeneration.
C9orf72 Repeat Expansion Pathology
The GGGGCC hexanucleotide repeat expansion in C9orf72 is the most common genetic cause of both familial and sporadic ALS. This expansion leads to disease through three non-mutually exclusive mechanisms: loss of C9orf72 protein function (haploinsufficiency), RNA toxicity from sense and antisense RNA foci that sequester RNA-binding proteins, and dipeptide repeat protein (DPR) toxicity from unconventional repeat-associated non-ATG (RAN) translation producing five different DPR species (poly-GA, poly-GP, poly-GR, poly-PA, poly-PR).
4. Etiology and Risk Factors
Genetic Factors
More than 40 genes have been associated with ALS. The most significant include:
- C9orf72 — hexanucleotide repeat expansion; accounts for 40% of familial and 7% of sporadic ALS cases; also associated with frontotemporal dementia
- SOD1 (superoxide dismutase 1) — the first ALS gene identified (1993); over 180 mutations known; accounts for approximately 12-20% of familial ALS
- TARDBP (TDP-43) — mutations account for approximately 4% of familial ALS
- FUS (fused in sarcoma) — mutations account for approximately 4% of familial ALS; often associated with younger onset
- UBQLN2 (ubiquilin 2) — X-linked dominant inheritance
- VCP (valosin-containing protein) — also causes inclusion body myopathy and Paget disease
- OPTN (optineurin) — associated with both ALS and glaucoma
- TBK1 (TANK-binding kinase 1) — involved in autophagy and neuroinflammation pathways
Environmental Risk Factors
- Cigarette smoking — the most consistently identified environmental risk factor; approximately 1.4-fold increased risk
- Military service — particularly Gulf War veterans; ALS is recognized as a service-connected disease by the U.S. Department of Veterans Affairs
- Heavy metal exposure — lead, mercury, and manganese have been implicated in some epidemiological studies
- Pesticide and herbicide exposure — organophosphates and other agricultural chemicals
- Head trauma — repeated traumatic brain injury, particularly in contact sports athletes
- Intense physical activity — professional athletes, particularly soccer players, show elevated risk in some studies, though the mechanism is debated
- Cyanobacterial toxins (BMAA) — beta-methylamino-L-alanine, proposed as the environmental factor in Western Pacific ALS clusters
Demographic Risk Factors
- Age — peak incidence between 55-75 years
- Sex — males at slightly higher risk (ratio approximately 1.3-1.5:1)
- Ethnicity — higher incidence in Caucasian populations compared to African, Asian, and Hispanic populations
- Family history — 5-10% of cases are familial with autosomal dominant inheritance being most common
5. Clinical Presentation
Upper Motor Neuron Signs
- Spasticity — velocity-dependent increase in muscle tone
- Hyperreflexia — exaggerated deep tendon reflexes
- Pathological reflexes — Babinski sign (extensor plantar response), Hoffman sign
- Pseudobulbar affect — involuntary, uncontrollable episodes of laughing or crying disproportionate to the emotional stimulus
- Clonus — rhythmic, involuntary muscular contractions
- Jaw jerk reflex — exaggerated in bulbar UMN involvement
Lower Motor Neuron Signs
- Muscle weakness — progressive, asymmetric, beginning focally and spreading contiguously
- Muscle atrophy — visible wasting, particularly in the intrinsic hand muscles (thenar and hypothenar eminences), forearms, and tongue
- Fasciculations — spontaneous, involuntary twitching of muscle fibers visible under the skin
- Hyporeflexia — diminished reflexes in severely atrophied muscles
- Muscle cramps — painful involuntary contractions, often an early symptom
- Flaccid weakness — decreased muscle tone in affected segments
Bulbar Symptoms
- Dysarthria — slurred, nasal, or strained speech; eventual anarthria
- Dysphagia — difficulty swallowing, choking on liquids, and progressive inability to manage oral secretions
- Tongue atrophy and fasciculations — a characteristic finding on examination
- Sialorrhea — drooling due to impaired swallowing of saliva rather than overproduction
- Laryngospasm — sudden vocal cord spasm causing stridor
Respiratory Symptoms
- Dyspnea on exertion — progressing to dyspnea at rest
- Orthopnea — difficulty breathing when lying flat due to diaphragmatic weakness
- Sleep-disordered breathing — nocturnal hypoventilation causing morning headaches and daytime somnolence
- Weak cough — impaired secretion clearance increasing pneumonia risk
Cognitive and Behavioral Changes
Up to 50% of ALS patients exhibit some degree of cognitive or behavioral impairment. Approximately 13-15% meet criteria for comorbid frontotemporal dementia (FTD), characterized by executive dysfunction, behavioral disinhibition, apathy, and language deficits. This ALS-FTD overlap is particularly common in patients carrying the C9orf72 repeat expansion.
6. Diagnosis
ALS remains a clinical diagnosis, as there is no single definitive diagnostic test. The average time from symptom onset to diagnosis is approximately 10-16 months, and patients often see multiple physicians before receiving the correct diagnosis.
Revised El Escorial Criteria (World Federation of Neurology)
The diagnosis requires the presence of:
- Evidence of lower motor neuron degeneration by clinical, electrophysiological, or neuropathological examination
- Evidence of upper motor neuron degeneration by clinical examination
- Progressive spread of symptoms within a region or to other regions
- Absence of electrophysiological or pathological evidence of other disease processes
Diagnostic certainty categories:
- Clinically definite ALS — UMN and LMN signs in three or more regions
- Clinically probable ALS — UMN and LMN signs in two regions with UMN signs rostral to LMN signs
- Clinically probable ALS, laboratory-supported — UMN and LMN signs in one region with EMG evidence of LMN involvement in two or more limbs
- Clinically possible ALS — UMN and LMN signs in one region, or UMN signs in two or more regions
Electrophysiological Studies
- Electromyography (EMG) — shows active and chronic denervation (fibrillation potentials, positive sharp waves, fasciculation potentials, large motor unit potentials with reduced recruitment) in clinically affected and unaffected muscles
- Nerve conduction studies (NCS) — motor and sensory nerve conduction velocities are typically normal or near-normal, helping to exclude peripheral neuropathies and multifocal motor neuropathy
Laboratory Studies
- Serum creatine kinase (CK) — mildly to moderately elevated in most patients
- Neurofilament light chain (NfL) — elevated in serum and CSF; increasingly used as a diagnostic and prognostic biomarker
- Genetic testing — recommended for patients with family history; screening panels available for C9orf72, SOD1, TARDBP, FUS, and other genes
- Exclusionary labs — thyroid function, vitamin B12, copper, parathyroid hormone, serum protein electrophoresis, anti-GM1 antibodies
Neuroimaging
- MRI of the brain and spinal cord — primarily used to exclude structural lesions, myelopathy, and other mimics; may show T2 hyperintensity along the corticospinal tracts
- PET and functional MRI — research tools showing motor cortex hypometabolism and altered connectivity
Pulmonary Function Testing
- Forced vital capacity (FVC) — baseline measurement and serial monitoring; FVC below 50% predicted indicates significant respiratory compromise
- Maximal inspiratory pressure (MIP) — more sensitive for early diaphragmatic weakness
- Sniff nasal inspiratory pressure (SNIP) — useful in patients with facial weakness who cannot form an adequate seal for standard spirometry
7. Treatment
ALS treatment is multidisciplinary and focuses on slowing disease progression, managing symptoms, maintaining function, and optimizing quality of life. Care is best delivered through specialized multidisciplinary ALS clinics, which have been shown to improve survival and quality of life.
Disease-Modifying Therapies
- Riluzole (Rilutek) — the first FDA-approved drug for ALS (1995); a glutamate antagonist that modestly extends survival by approximately 2-3 months; dose is 50 mg twice daily; available in oral tablet and liquid formulation (Tiglutik)
- Edaravone (Radicava) — FDA-approved in 2017; a free radical scavenger that slows functional decline by approximately 33% as measured by the ALS Functional Rating Scale-Revised (ALSFRS-R) in a select patient population; administered intravenously or orally
- Sodium phenylbutyrate-taurursodiol (Relyvrio/AMX0035) — FDA-approved in 2022; targets endoplasmic reticulum stress and mitochondrial dysfunction; shown to slow functional decline in clinical trials
- Tofersen (Qalsody) — FDA-approved in 2023 for SOD1-ALS; an antisense oligonucleotide (ASO) that reduces SOD1 protein production; administered intrathecally
Symptom Management
- Spasticity — baclofen, tizanidine, benzodiazepines, dantrolene
- Sialorrhea — glycopyrrolate, atropine sublingual drops, botulinum toxin injections to salivary glands, suction devices
- Pseudobulbar affect — dextromethorphan-quinidine (Nuedexta), the only FDA-approved treatment for this symptom
- Pain — NSAIDs, gabapentin, pregabalin, opioids for refractory cases
- Depression and anxiety — SSRIs, SNRIs, mirtazapine
- Muscle cramps — mexiletine, quinine (limited evidence), magnesium
- Insomnia — trazodone, zolpidem, melatonin
- Constipation — stool softeners, stimulant laxatives, adequate hydration
Respiratory Support
- Non-invasive ventilation (NIV/BiPAP) — recommended when FVC falls below 50% or symptoms of nocturnal hypoventilation develop; extends survival by a median of 7-13 months and significantly improves quality of life
- Mechanical insufflation-exsufflation (cough assist) — helps clear secretions and reduces pneumonia risk
- Invasive mechanical ventilation (tracheostomy) — considered when NIV is no longer sufficient; can sustain life for years but requires 24-hour care; chosen by approximately 5-10% of ALS patients in the United States
- Diaphragm pacing — no longer recommended based on negative clinical trial results
Nutritional Support
- Dietary modifications — high-calorie, high-protein diet; modified food textures for dysphagia
- Percutaneous endoscopic gastrostomy (PEG) — recommended when swallowing becomes unsafe or weight loss exceeds 10% of baseline; stabilizes weight and simplifies medication administration
- Radiologically inserted gastrostomy (RIG) — alternative for patients with low FVC who may not tolerate sedation for PEG placement
Rehabilitation and Assistive Devices
- Physical therapy — gentle range-of-motion exercises, stretching, and adaptive exercise programs
- Occupational therapy — adaptive equipment, home modifications, energy conservation strategies
- Speech therapy — communication strategies, voice banking, augmentative and alternative communication (AAC) devices, eye-tracking technology
- Assistive technology — ankle-foot orthoses (AFOs), wheelchairs, hospital beds, environmental control units
8. Complications
- Respiratory failure — the leading cause of death in ALS; progressive diaphragmatic and intercostal muscle weakness
- Aspiration pneumonia — resulting from dysphagia and impaired cough reflex
- Malnutrition and cachexia — due to dysphagia, increased metabolic demands, and hypermetabolism
- Venous thromboembolism — due to immobility; deep vein thrombosis and pulmonary embolism
- Pressure ulcers — from immobility and malnutrition
- Falls and fractures — resulting from progressive weakness and impaired balance
- Depression and anxiety — affecting up to 44% of patients
- Social isolation — due to communication difficulties and mobility limitations
- Caregiver burden — significant psychological, physical, and financial strain on family caregivers
- Frontotemporal dementia — cognitive and behavioral decline complicating care decisions and advance planning
- Laryngospasm — sudden airway obstruction causing significant patient distress
- Pain syndromes — musculoskeletal pain, joint contractures, spasticity-related pain
9. Prognosis
ALS is invariably fatal, with a median survival of 2-5 years from symptom onset. Approximately 50% of patients die within 30 months of symptom onset, and approximately 80-90% die within 5 years. However, about 5-10% of patients survive beyond 10 years, and rare individuals survive 20 years or more, as exemplified by physicist Stephen Hawking, who lived 55 years after diagnosis.
Factors associated with poorer prognosis include:
- Bulbar-onset disease — median survival approximately 2-2.5 years
- Older age at onset — particularly onset after age 75
- Rapid progression — significant decline in ALSFRS-R score in the first 3 months
- Low FVC at diagnosis — particularly below 70% predicted
- Comorbid frontotemporal dementia — reduces median survival by approximately 6-12 months
- Low BMI and weight loss — malnutrition is an independent predictor of shorter survival
- Diagnostic delay — longer delay paradoxically correlates with slower progression (selection bias)
Factors associated with better prognosis include:
- Younger age at onset — particularly onset before age 40
- Limb-onset disease — median survival approximately 3-5 years
- Predominantly LMN phenotype (progressive muscular atrophy)
- Flail arm or flail leg variants — slower progression phenotypes
- Multidisciplinary clinic care — associated with 7-10 months longer survival
- Early use of NIV — extends survival by a median of 7-13 months
10. Prevention
There are no proven strategies to prevent ALS, as the etiology in most cases remains unknown. However, modifiable risk factors that may reduce risk include:
- Smoking cessation — smoking is the most consistently identified modifiable risk factor
- Minimizing toxic exposures — reducing occupational exposure to heavy metals, pesticides, and industrial chemicals
- Head injury prevention — use of helmets and safety equipment in sports and occupational settings
- Genetic counseling — for family members of individuals with familial ALS; presymptomatic genetic testing is available for known ALS genes
For individuals carrying known ALS gene mutations, presymptomatic monitoring with serial neurological examinations, neurofilament light chain levels, and electrophysiological studies may allow earlier detection and future access to gene-targeted therapies such as tofersen for SOD1 carriers.
Interactive Visualization Protein Folding — fold it right, or watch it clump Watch a chaperone help a new protein fold into shape — then take it away and see it clump into the aggregates behind Alzheimer's and Huntington's, or a prion convert its neighbours. Launch →
Table of Contents
- Overview
- Epidemiology
- Pathophysiology
- Etiology and Risk Factors
- Clinical Presentation
- Diagnosis
- Treatment
- Complications
- Prognosis
- Prevention
- Research Papers
- Connections
- Featured Videos
Research Papers
Curated PubMed topic searches on ALS (Amyotrophic Lateral Sclerosis). Each link opens a live PubMed query so the result set stays current as new studies are indexed.
- PubMed topic search: Amyotrophic lateral sclerosis review
- PubMed topic search: ALS pathophysiology
- PubMed topic search: SOD1 gene ALS
- PubMed topic search: C9orf72 ALS FTD
- PubMed topic search: TDP-43 ALS
- PubMed topic search: Riluzole ALS trial
- PubMed topic search: Edaravone ALS trial
- PubMed topic search: Tofersen SOD1 ALS
- PubMed topic search: ALS El Escorial criteria
- PubMed topic search: ALS multidisciplinary clinic outcomes
- PubMed topic search: ALS non-invasive ventilation
- PubMed topic search: ALS neurofilament biomarker
Connections
- How Proteins Fold — and Misfold in Disease — interactive animation
- Magnesium
- Vitamin B12
- Multiple Sclerosis
- Parkinson's Disease
- Huntington's Disease
- Myasthenia Gravis
- Peripheral Neuropathy
- Lion's Mane Mushroom
- Dementia
- Stress Management
- Oxidative Stress
- Heavy Metals
- Lead
- Creatine
- Alzheimer's Disease
- Depression
- Numbness Tingling