Hepatic Encephalopathy
Hepatic encephalopathy (HE) is a brain dysfunction caused by liver insufficiency and/or portosystemic shunting of blood, allowing neurotoxins — most critically ammonia — to bypass hepatic detoxification and reach the central nervous system. The resulting spectrum of neurological and psychiatric disturbances ranges from subtle cognitive impairment detectable only by psychometric testing (covert or minimal HE) to profound disorientation, asterixis, stupor, and life-threatening coma (overt HE grades 2–4). HE complicates both chronic liver disease — particularly cirrhosis, where 30–45% of patients develop at least one overt episode — and acute liver failure (ALF), where cerebral edema and raised intracranial pressure can cause fatal herniation. Recurrent overt HE is associated with irreversible neurocognitive damage, loss of employment, inability to drive, and sharply reduced quality of life. Effective management hinges on identifying and reversing precipitating factors, reducing intestinal ammonia production (lactulose, rifaximin), and supporting the patient through acute decompensation.
Table of Contents
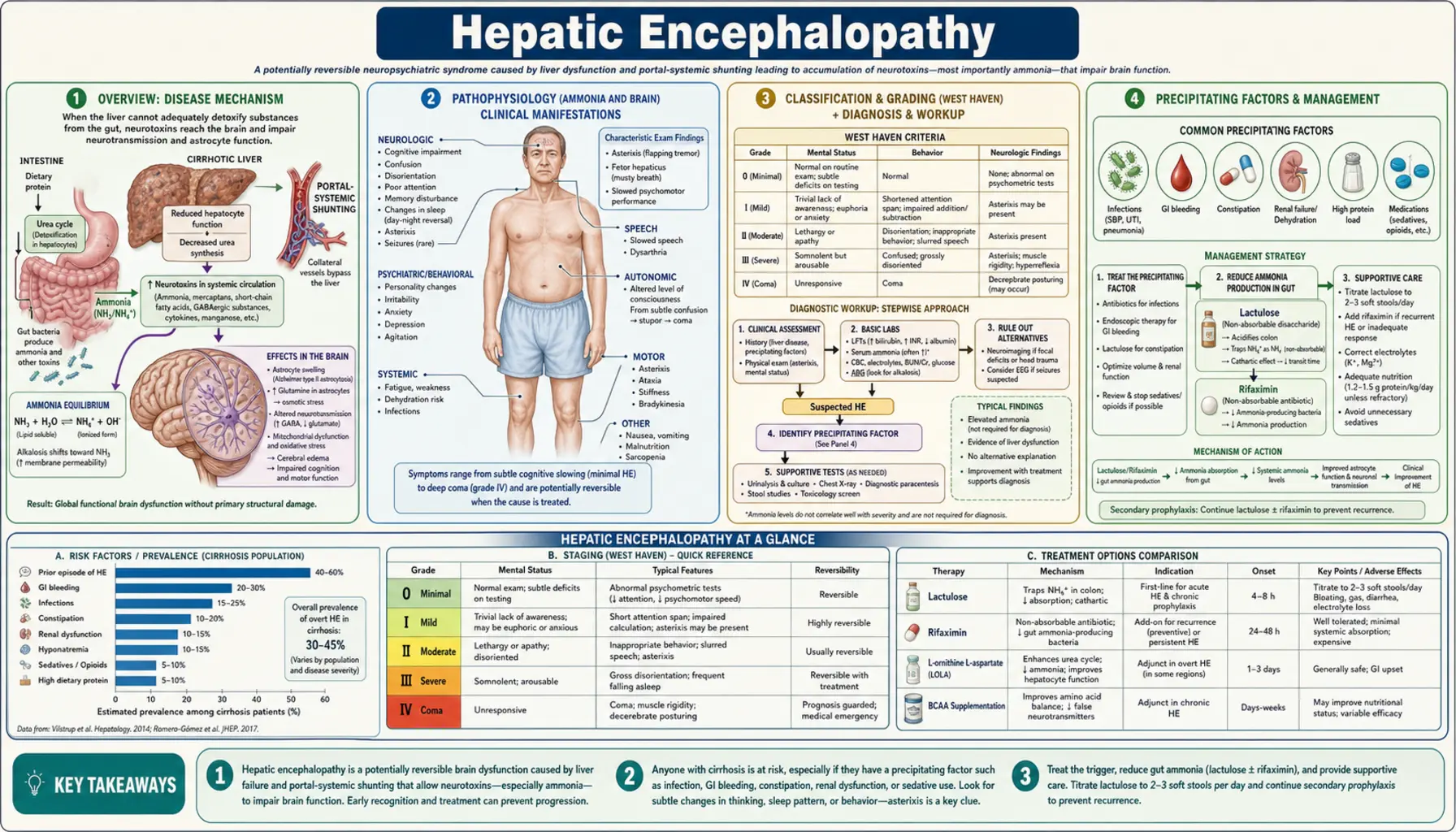
- Overview
- Pathophysiology (Ammonia and Brain)
- Classification and Grading (West Haven)
- Precipitating Factors
- Clinical Presentation
- Diagnosis
- Lactulose Therapy
- Rifaximin and Other Treatments
- TIPS-Associated Encephalopathy
- Nutrition and Supportive Care
- References
Overview
Hepatic encephalopathy is defined as the brain dysfunction arising directly from liver insufficiency and/or portosystemic shunting, after excluding other known brain diseases. It is classified along a continuum: covert HE (grades 0–1, formerly minimal HE) produces no obvious behavioral change but impairs performance on psychometric tests, reduces driving safety, and undermines work and social function; overt HE (grades 2–4) involves clinically apparent disorientation, personality change, asterixis, and, at the extreme, unresponsive coma.
HE occurs in 30–45% of patients with cirrhosis over their disease course. A single overt episode marks a turning point: one-year survival falls to roughly 40%, and each recurrence accelerates cognitive decline. Patients with recurrent or persistent overt HE accumulate lasting neurocognitive damage even between episodes — a phenomenon termed post-HE syndrome.
In acute liver failure, the pathophysiology differs in critical ways: cerebral edema rather than neuroinhibition dominates, raised intracranial pressure can cause fatal transtentorial herniation, and ammonia levels tend to be far higher than in cirrhosis. Management of ALF-related HE requires intensive care with airway protection, ICP monitoring in grade 3–4, and urgent evaluation for emergency liver transplantation.
Although ammonia is the central neurotoxin shared across all HE subtypes, it does not act alone. Systemic inflammation, gut microbiome dysbiosis, oxidative stress, and zinc deficiency all lower the threshold at which ammonia produces encephalopathy, explaining why seemingly similar ammonia levels produce wildly different clinical severity in different patients.
Pathophysiology (Ammonia and Brain)
Under normal conditions, intestinal bacteria and enterocytes generate ammonia (NH3) from dietary protein, glutamine, and urea hydrolysis. The portal blood carries this ammonia to the liver, where the urea cycle (ornithine transcarbamylase, carbamoyl phosphate synthetase, and downstream enzymes) converts it to urea, which is then excreted renally. In cirrhosis, two mechanisms disrupt this clearance: (1) hepatocyte mass and urea cycle enzyme capacity are severely reduced, and (2) portosystemic shunts — both spontaneous collaterals and surgically placed TIPS — bypass the liver entirely, delivering ammonia directly into the systemic circulation.
Ammonia crosses the blood-brain barrier readily. Astrocytes, the only brain cells that express glutamine synthetase, take up ammonia and detoxify it by incorporating it into glutamine. The resulting cerebral glutamine accumulation acts as an osmolyte, causing astrocyte swelling and cytotoxic cerebral edema (the glutamine osmotic hypothesis). Swollen astrocytes impair synaptic glutamate recycling and neurotransmitter homeostasis, disrupting neural signaling broadly.
Additional neurotoxins amplify ammonia's effect:
- Manganese: Inadequately cleared by the diseased liver, manganese deposits in the basal ganglia (globus pallidus), producing T1-hyperintensity on MRI and Parkinsonian features. It potentiates ammonia-induced astrocyte swelling.
- Inflammatory cytokines (IL-6, TNF-α): Systemic inflammation — from spontaneous bacterial peritonitis, infections, or gut translocation — dramatically synergizes with ammonia. The SONIC study demonstrated that patients with both elevated ammonia and systemic inflammation had far worse cognitive performance than patients with either alone.
- Reactive oxygen and nitrogen species (ROS/RNS): Glutamine hydrolysis inside mitochondria re-generates ammonia intracellularly, producing oxidative stress and mitochondrial dysfunction in astrocytes.
- GABA-A receptor upregulation: Increased neurosteroid (allopregnanolone) production and benzodiazepine-like ligands enhance inhibitory GABAergic neurotransmission, contributing to sedation and coma.
The gut microbiome is a critical upstream driver. Urease-producing bacteria — notably Proteus mirabilis and Helicobacter pylori — hydrolyze urea to ammonia in the colon. Cirrhosis-associated dysbiosis (reduced Lachnospiraceae, expanded Enterobacteriaceae) amplifies colonic ammonia generation and gut mucosal permeability, facilitating systemic endotoxemia and inflammation. Zinc deficiency, extremely common in cirrhosis, further impairs urea cycle enzyme activity, reducing hepatic ammonia clearance capacity even in residual functional hepatocytes.
Classification and Grading (West Haven)
The West Haven Criteria remain the most widely used clinical grading system for overt HE:
- Grade 0 (Covert / Minimal HE): No detectable personality or behavioral change on clinical examination. Abnormalities are identified only by neuropsychometric or neurophysiological testing. Despite the absence of obvious symptoms, covert HE impairs driving performance, occupational productivity, and health-related quality of life. Prevalence in stable outpatient cirrhosis is 20–80% depending on detection method.
- Grade 1 (Covert): Trivial lack of awareness; euphoria or anxiety; shortened attention span; impaired performance of addition or subtraction. Subtle personality changes may be noticed by family members before the clinician.
- Grade 2 (Overt): Lethargy or apathy; disorientation to time; obvious personality change; inappropriate behavior; asterixis (flapping tremor of outstretched hands). Patients are functional but require supervision.
- Grade 3 (Overt): Somnolence to semi-stupor; responsive to verbal stimuli; gross disorientation; bizarre behavior. Patients cannot care for themselves.
- Grade 4 (Overt): Coma — unresponsive to verbal or painful stimuli, with or without decerebrate posturing.
The International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN) classification overlays clinical course on grade: episodic (single precipitated or spontaneous episode), recurrent (two or more episodes within 6 months), and persistent (behavioral alterations always present, interspersed with relapses). This distinction matters for treatment decisions — rifaximin is specifically approved and most cost-effective in recurrent overt HE.
For covert HE detection, several validated instruments are available: the Psychometric Hepatic Encephalopathy Score (PHES — a battery of 5 paper-and-pencil tests), the Stroop smartphone application (EncephalApp), and the Critical Flicker Frequency (CFF) test. These tools are underutilized in clinical practice but identify patients at high risk of progressing to overt HE who may benefit from early intervention.
Precipitating Factors
Identifying and correcting the precipitating factor is as critical as treating the HE itself. In 80–90% of cases of overt HE in cirrhosis, a trigger can be identified:
- Gastrointestinal bleeding: The most potent precipitant. Blood in the GI tract represents a massive nitrogen load — approximately 10 g of protein per 100 mL of blood — which is rapidly metabolized by gut bacteria to ammonia. Even a small variceal bleed can precipitate grade 3–4 HE within hours.
- Infection: Spontaneous bacterial peritonitis (SBP), urinary tract infection, and pneumonia trigger systemic inflammation that lowers the HE threshold (SONIC study). Every cirrhotic patient with acute HE must be screened for occult infection with blood cultures, urinalysis, and diagnostic paracentesis.
- Constipation: Prolonged transit time increases colonic bacterial ammonia production. Adequate bowel frequency (2–3 stools/day) is a cornerstone of prevention.
- Electrolyte disturbances: Hypokalemia drives increased renal ammoniagenesis (potassium depletion stimulates renal glutaminase). Hyponatremia impairs cerebral osmoregulation, synergizing with glutamine-induced astrocyte swelling. Both are common in cirrhosis due to diuretic use and aldosterone excess.
- Diuretic overuse: Excessive furosemide or spironolactone causes hypovolemia, pre-renal azotemia, alkalosis (which favors NH3 over NH4+, increasing cerebral penetration), and hypokalemia — a perfect storm for HE precipitation.
- Sedatives, opioids, and benzodiazepines: These enhance GABAergic neurotransmission directly, dramatically potentiating the neuroinhibitory effects of ammonia and other HE neurotoxins. Even standard doses can precipitate severe HE in advanced cirrhosis.
- Hepatocellular carcinoma (HCC) development or progression: Tumor growth displaces functional liver mass and may cause portal vein thrombosis, worsening shunting.
- Dehydration: Reduces renal perfusion, increases urea recycling, and concentrates ammonia.
- TIPS procedure: Deliberate creation of a large portosystemic shunt is itself a major precipitant (see Section 9).
- Non-adherence to lactulose or rifaximin: One of the most common causes of recurrent overt HE in outpatients. Simplifying regimens, using liquid lactulose with dose titration, and addressing cost barriers for rifaximin are essential.
Clinical Presentation
The clinical features of HE span a wide spectrum and evolve as encephalopathy grade worsens:
- Sleep-wake cycle reversal: One of the earliest and most consistent signs — patients become drowsy during the day and awake and agitated at night. This often precedes obvious cognitive symptoms and reflects disruption of circadian melatonin regulation.
- Personality and behavioral changes: Irritability, apathy, disinhibition, and mood lability. Family members typically notice these changes before the clinician does; directed questioning of caregivers is essential.
- Asterixis (flapping tremor): The pathognomonic motor sign of metabolic encephalopathy. Elicited by asking the patient to extend arms and dorsiflex wrists for 15 seconds — rhythmic, arrhythmic lapses of sustained posture produce the characteristic flap. Present in grades 2–3; absent in grade 4 (deep coma) and in covert HE.
- Fetor hepaticus: A sweet, musty, slightly feculent breath odor caused by pulmonary excretion of dimethyl sulfide, a byproduct of bacterial methionine metabolism. When present, it is virtually diagnostic of significant portosystemic shunting.
- Constructional apraxia: Inability to reproduce simple geometric figures (e.g., a 5-pointed star or interlocking pentagons). A quick bedside test sensitive for grades 1–2 HE.
- Slurred speech and psychomotor slowing: Bradyphrenia (slow thinking) and dysarthria reflect global cerebral depression.
- Parkinsonian features: Resting tremor, cogwheel rigidity, and bradykinesia in patients with chronic, recurrent HE — attributed to manganese deposition in the globus pallidus and substantia nigra.
- Driving impairment: Patients with even covert HE have reaction times and hazard detection abilities comparable to legally intoxicated drivers. Physicians should counsel patients about driving restrictions.
- In acute liver failure: Cerebral edema dominates — papilledema, decerebrate posturing, Cushing's triad (bradycardia, hypertension, irregular respirations), and ultimately brainstem herniation if untreated. This presentation requires emergent neurocritical care.
Diagnosis
HE is primarily a clinical diagnosis supported by the presence of liver disease or portosystemic shunting, compatible neurological features, and exclusion of alternative causes. There is no single laboratory test that confirms or rules out HE.
Serum ammonia: Elevated in most patients with overt HE (venous ammonia >100–150 µmol/L is supportive), but the correlation between ammonia level and clinical grade is poor — particularly in patients with muscle wasting (reduced peripheral ammonia uptake) or those on norfloxacin. A normal ammonia level does not exclude HE; an elevated level in an alert patient does not establish it. In ALF, arterial ammonia (>200 µmol/L) correlates with risk of intracranial hypertension and herniation and is used to guide ICP monitoring decisions. Ammonia must be measured in an ice-cooled tube processed within 15 minutes.
Neuroimaging (CT/MRI): Essential to exclude structural causes of altered mental status — subdural hematoma (especially in thrombocytopenic cirrhotic patients prone to falls), ischemic or hemorrhagic stroke, and Wernicke's encephalopathy. MRI with T1 sequences may reveal symmetric pallidal hyperintensity from manganese deposition — a marker of chronic portosystemic shunting rather than acute HE severity.
Electroencephalography (EEG): Shows characteristic triphasic waves (bifrontally predominant, 1.5–3 Hz) in moderate-to-severe HE. Useful for excluding non-convulsive status epilepticus, which can mimic HE. EEG changes generally parallel clinical grade but are not specific to HE.
Psychometric tests for covert HE: The PHES (5-test paper battery: number connection A and B, digit-symbol coding, serial dotting, line-tracing) is the reference standard. EncephalApp Stroop is validated and can be administered in minutes on a smartphone. Critical Flicker Frequency (CFF <39 Hz) is abnormal in covert HE. These tests identify patients who warrant prophylactic therapy and are unfit to drive.
Portosystemic shunt mapping: CT angiography or MRA identifies large spontaneous splenorenal or gastrorenal shunts that can be embolized in selected patients with refractory HE despite maximal medical therapy.
Lactulose Therapy
Lactulose is a synthetic, non-absorbable disaccharide (galactose-fructose) and the first-line treatment for both acute overt HE and secondary prevention of recurrence. Standard dosing for acute episodes is 30–45 mL orally every 4–6 hours, titrated to achieve 2–3 soft stools per day. For patients unable to take lactulose orally (grade 3–4 HE, ileus), retention enemas (300 mL lactulose in 700 mL water, retained for 30–60 minutes, 3–4 times daily) are effective.
Mechanisms of action:
- Colonic acidification: Gut bacteria ferment lactulose to short-chain fatty acids (acetate, propionate, butyrate), reducing colonic pH from ~7 to ~5. At lower pH, NH3 is protonated to NH4+, which is membrane-impermeant and thus trapped in the stool rather than absorbed — the ionic trapping mechanism.
- Microbiome modulation: Acidic pH and fermentation substrate shift favor saccharolytic over proteolytic/urease-producing bacteria, reducing ammonia production.
- Cathartic effect: Accelerated transit reduces intestinal contact time for ammonia absorption.
Secondary prevention: After a first overt HE episode, lactulose maintenance therapy reduces recurrence by approximately 50% (Sharma et al., Hepatology 2009). Guidelines from EASL/AASLD recommend indefinite lactulose for all patients who survive an overt episode.
Side effects and pitfalls: Diarrhea, flatulence, bloating, and hypernatremia from excess free-water stool losses are common dose-dependent side effects. Overtreatment is a frequent and underappreciated cause of worsening HE — excessive diarrhea causes dehydration, electrolyte disturbances (hyponatremia, hypokalemia), and pre-renal azotemia, all of which precipitate or deepen encephalopathy. Patients and caregivers must be educated that the target is 2–3 soft stools per day, not maximum frequency.
Rifaximin and Other Treatments
Rifaximin is a minimally absorbed (<0.4% systemic bioavailability), broad-spectrum rifamycin antibiotic that acts locally in the GI tract to suppress urease-producing bacteria and reduce intestinal ammonia generation. The pivotal NEJM 2010 trial (Bass et al.) randomized 299 patients with a history of overt HE to rifaximin 550 mg twice daily or placebo for 6 months. Rifaximin reduced breakthrough overt HE episodes by 58% (22.1% vs. 45.9%) and HE-related hospitalizations by 50%, both on a background of lactulose use in ~90% of participants. Rifaximin received FDA approval in 2010 for maintenance and secondary prevention of overt HE in adults with cirrhosis.
Combination therapy: A randomized trial by Sharma et al. (2013) demonstrated that rifaximin plus lactulose was superior to lactulose alone for acute overt HE (complete reversal: 76% vs. 44%; mortality: 24% vs. 49%). Combination therapy is now recommended for moderate-to-severe acute overt HE and for secondary prevention in patients who fail lactulose monotherapy.
Cost and access: Rifaximin (Xifaxan) costs approximately $1,100–$1,400/month in the United States, creating substantial access barriers. Generic rifaximin became available in the US in 2024, improving affordability. International availability varies widely.
Other pharmacological approaches:
- L-Ornithine L-Aspartate (LOLA): Provides substrates for the urea cycle and glutamine synthesis, increasing hepatic and skeletal muscle ammonia detoxification. IV LOLA reduces ammonia levels and improves HE grade in RCTs (Kircheis et al., 1997). Widely used in Europe, India, and Latin America but not FDA-approved in the US.
- Zinc supplementation: Zinc 220 mg sulfate twice daily corrects the near-universal zinc deficiency in cirrhosis and restores urea cycle enzyme function. Safe, inexpensive, and recommended by AASLD guidelines for zinc-deficient patients with HE.
- Albumin infusions: Albumin binds and neutralizes neurotoxins in the bloodstream. Long-term albumin infusion (10 g/day weekly) is under investigation in the PILOT study and several European trials; early results suggest reduction in HE and other decompensations.
- Sodium benzoate: An alternative ammonia scavenger that conjugates glycine to form hippurate (renally excreted), bypassing the urea cycle. Randomized trial by Sushma et al. (1992) showed comparable efficacy to lactulose at lower cost. Used as a second-line or cost-saving alternative in some centers.
- Branched-chain amino acids (BCAAs): BCAA supplementation (leucine, isoleucine, valine) corrects the reduced plasma BCAA:aromatic amino acid ratio in cirrhosis, potentially improving encephalopathy grade and quality of life. Most beneficial in protein-intolerant patients or those with severe sarcopenia.
- Fecal Microbiota Transplantation (FMT): Early-phase trials by Bajaj et al. demonstrated that FMT from a healthy donor improved cognitive function, reduced hospitalizations, and shifted the gut microbiome toward a less ammonia-producing composition in cirrhotic patients with recurrent HE. Larger controlled trials are ongoing.
- Agents to avoid: Neomycin (nephrotoxic, ototoxic — use only as short-term bridge in resource-limited settings). Metronidazole (peripheral neuropathy and hepatotoxicity with prolonged use). Benzodiazepines (potentiate GABA inhibition — use with extreme caution if at all).
TIPS-Associated Encephalopathy
Transjugular intrahepatic portosystemic shunt (TIPS) placement creates a direct communication between the portal and hepatic veins, markedly reducing portal pressure. While lifesaving for refractory variceal bleeding and refractory ascites, TIPS dramatically increases portosystemic shunting — bypassing hepatic ammonia clearance — and precipitates new or worsened HE in 10–50% of patients within the first year post-procedure.
Risk factors for TIPS-induced HE: Age >65 years; Child-Pugh score C or MELD >18; pre-existing HE (even covert); low serum albumin (<2.5 g/dL); sarcopenia; prior spontaneous HE; large shunt diameter (10 mm vs. 8 mm).
Prevention strategies: Use of 8 mm covered stents rather than 10 mm reduces HE incidence (REDUCE trial) while maintaining adequate portal decompression for most indications. Patients should be started on or have lactulose and rifaximin optimized before TIPS placement whenever possible.
Management of established TIPS-induced HE:
- First-line: optimize lactulose (2–3 stools/day) + rifaximin 550 mg twice daily.
- Nutritional optimization: protein intake 1.2–1.5 g/kg/day, late-evening snack, BCAA supplements if protein-intolerant.
- For refractory HE unresponsive to maximal medical therapy: TIPS reduction (insertion of a constrictive ring or covered stent within the existing stent to reduce shunt diameter, typically from 10 to 8 mm) or TIPS occlusion. This must be weighed against the risk of re-accumulation of ascites or recurrent variceal bleeding.
Elective early TIPS (within 72 hours of variceal bleed) in high-risk patients (Child-Pugh B with active bleeding, or Child-Pugh C) carries an acceptable HE rate given its dramatic mortality benefit (NEJM 2010 García-Pagán et al.), but post-TIPS HE prophylaxis is mandatory.
Nutrition and Supportive Care
Protein restriction is harmful and must be abandoned. Decades of misguided practice restricted dietary protein in HE patients to reduce ammonia substrate. RCT evidence (Cordoba et al., 2004) demonstrated that normal protein intake during acute episodic HE produced no worse encephalopathy than protein restriction, while restricting protein worsened sarcopenia. Sarcopenia — ubiquitous in advanced cirrhosis — is itself an independent risk factor for HE, because skeletal muscle is the main extrahepatic ammonia detoxification organ (via glutamine synthetase). The EASL/AASLD guidelines now recommend:
- Protein intake: 1.2–1.5 g/kg/day (based on ideal or dry body weight) in all grades of HE.
- Small, frequent meals (4–6 per day) rather than 2–3 large meals, to minimize fasting-induced catabolism.
- Late-evening snack (complex carbohydrates + protein, 200–400 kcal): reduces the overnight fasting period, suppresses gluconeogenic catabolism of muscle protein, and reduces morning ammonia generation. This single intervention has shown HE-prevention benefit in RCTs.
- Vegetable and dairy protein preferred over red meat protein: plant proteins are less ammoniagenic (lower methionine and aromatic amino acid content, higher fiber promoting stool excretion of nitrogen), and dairy protein provides BCAAs without the same ammonia burden.
- BCAA-enriched supplements for patients who are genuinely protein-intolerant (nausea, encephalopathy worsening at standard doses) — can improve HE grade and allow adequate nitrogen intake.
Acute grade 3–4 HE and ICU management:
- Airway protection: Intubation threshold is low — aspiration risk from loss of airway reflexes, and need for endotracheal intubation before procedures (paracentesis, endoscopy) in obtunded patients.
- Sedation: Minimize all sedatives. If procedural sedation is required, propofol (short-acting, no GABA enhancement at low doses) or dexmedetomidine is preferred over benzodiazepines.
- Opioid use: If analgesics are unavoidable, short-acting fentanyl is preferred; morphine has active metabolites that accumulate in renal impairment; avoid codeine.
- ICP monitoring and cerebral edema: In ALF with grade 3–4 HE, arterial ammonia >200 µmol/L, or any clinical sign of raised ICP — mannitol (0.5–1 g/kg IV bolus), 3% hypertonic saline to maintain Na 145–155 mEq/L, and head of bed elevation to 30°. Therapeutic hypothermia to 33–35°C reduces ICP and ammonia in severe ALF.
- Liver transplantation: The definitive treatment for recurrent or persistent refractory HE in decompensated cirrhosis. Post-transplant, most patients experience substantial cognitive recovery; a subset with severe pre-transplant neurocognitive damage has persistent deficits. HE is a recognized MELD exception criterion at many transplant centers.
References
- Bass NM, Mullen KD, Sanyal A, et al. Rifaximin treatment in hepatic encephalopathy. N Engl J Med. 2010;362(12):1071-1081. — PMID: 20335583
- Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 practice guideline by the European Association for the Study of the Liver and the American Association for the Study of Liver Diseases. J Hepatol. 2014;61(3):642-659. — PMID: 25015420
- Bajaj JS, Kamath PS, Reddy KR. The evolving challenge of infections in cirrhosis. N Engl J Med. 2021;384(24):2317-2330. — Search PubMed
- Cordoba J, Lopez-Hellin J, Planas M, et al. Normal protein diet for episodic hepatic encephalopathy: results of a randomized study. J Hepatol. 2004;41(1):38-43. — PMID: 15246205
- Mullen KD, Sanyal AJ, Bass NM, et al. Rifaximin is safe and well tolerated for long-term maintenance of remission from overt hepatic encephalopathy. Clin Gastroenterol Hepatol. 2014;12(8):1390-1397. — PMID: 24365449
- Montagnese S, Bajaj JS. Impact of hepatic encephalopathy in cirrhosis on quality-of-life issues. Drugs. 2019;79(Suppl 1):11-16. — Search PubMed
- Bajaj JS, Heuman DM, Hylemon PB, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol. 2014;60(5):940-947. — Search PubMed
- Sharma BC, Sharma P, Lunia MK, et al. A randomized, double-blind, controlled trial comparing rifaximin plus lactulose with lactulose alone in treatment of overt hepatic encephalopathy. Am J Gastroenterol. 2013;108(9):1458-1463. — PMID: 23877348
- Amodio P, Del Piccolo F, Marchetti P, et al. Clinical features and survivorship of covert hepatic encephalopathy in patients with cirrhosis. Gastroenterology. 1999;117(6):1422-1430. — Search PubMed
- Kircheis G, Nilius R, Held C, et al. Therapeutic efficacy of L-ornithine-L-aspartate infusions in patients with cirrhosis and hepatic encephalopathy. Hepatology. 1997;25(6):1351-1360. — Search PubMed
- Tranah TH, Vijay GKM, Ryan JM, Shawcross DL. Systemic inflammation and ammonia in hepatic encephalopathy. Metab Brain Dis. 2013;28(1):1-5. — Search PubMed
- Bajaj JS, Fagan A, Gavis EA, et al. Long-term outcomes of fecal microbiota transplantation in patients with cirrhosis. Gastroenterology. 2019;156(6):1921-1923. — Search PubMed
- Featured Videos
Research Papers
- Hepatic encephalopathy and ammonia — PubMed
- Rifaximin for hepatic encephalopathy — PubMed
- Lactulose therapy in HE — PubMed
- West Haven grading criteria — PubMed
- TIPS-associated encephalopathy — PubMed
- Gut microbiome and HE in cirrhosis — PubMed
- Covert HE and driving impairment — PubMed
- FMT for hepatic encephalopathy — PubMed
Connections
- Lactulose — Ammonia, Gut Bacteria & the Liver (In-Depth)
- Cirrhosis
- Liver Disease
- Primary Biliary Cholangitis
- Non-Alcoholic Fatty Liver Disease
- Acute Kidney Injury
- Encephalopathy
- Hepatitis C
- Hepatitis B
- Zinc
- Glutamine
- Liver Cancer
- Ammonia Lab Test
- Lactulose — first-line therapy for overt HE and for preventing recurrence.