Primary Biliary Cholangitis
Primary biliary cholangitis (PBC) is a chronic, progressive, cholestatic autoimmune liver disease caused by immune-mediated destruction of the small intrahepatic bile ducts. Formerly called "primary biliary cirrhosis," the name was officially changed in 2015 to better reflect the disease's autoimmune nature and to reduce stigma — most patients do not have cirrhosis at diagnosis. The hallmark laboratory finding is the anti-mitochondrial antibody (AMA), specifically the M2 subtype targeting the E2 subunit of the pyruvate dehydrogenase complex, which is positive in approximately 95% of patients. PBC disproportionately affects women, with a female-to-male ratio of approximately 9:1, and onset typically occurs between the ages of 40 and 60. Without treatment, the disease progresses insidiously over years to decades, ultimately leading to biliary cirrhosis and liver failure. PBC frequently coexists with other autoimmune conditions including Sjögren's syndrome, autoimmune thyroid disease, and Raynaud's phenomenon.
Table of Contents
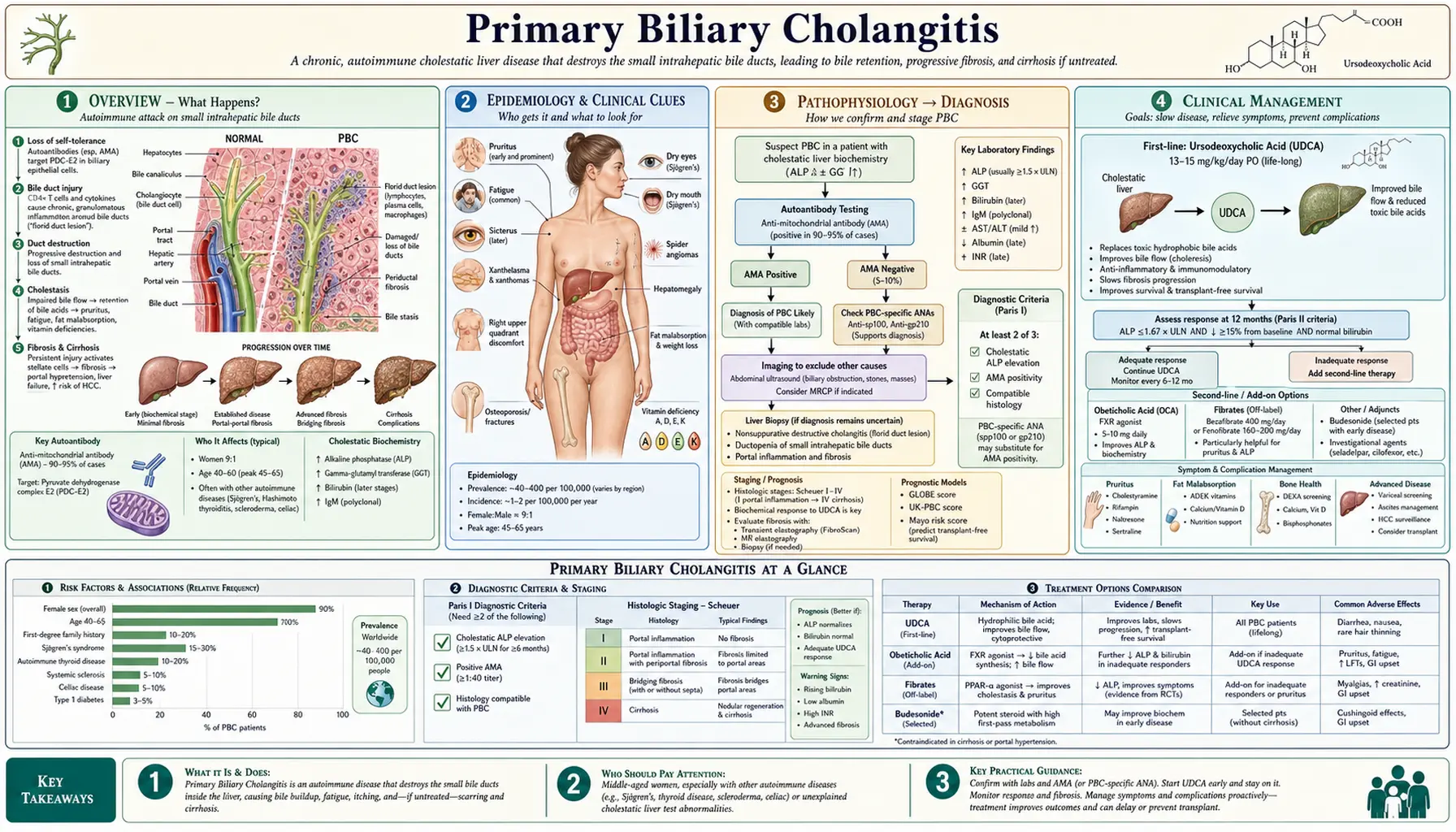
- Overview
- Epidemiology
- Pathophysiology
- Clinical Presentation
- Diagnosis and Staging
- Treatment — UDCA
- Second-Line Therapies
- Complications
- Prognosis
- References
1. Overview
Primary biliary cholangitis is a chronic cholestatic autoimmune liver disease characterized by immune-mediated targeting and progressive destruction of the small intrahepatic bile ducts (interlobular and septal bile ducts). This ductal destruction impairs bile flow (cholestasis), causing bile acids and other toxic biliary compounds to accumulate in the liver and bloodstream, gradually driving hepatic fibrosis and, in advanced cases, cirrhosis.
The disease was renamed from "primary biliary cirrhosis" to "primary biliary cholangitis" in 2015 following a patient-led advocacy effort, as the word "cirrhosis" carries social stigma and was inaccurate for the majority of patients who present at an earlier, non-cirrhotic stage. The new name reflects the core pathological process — cholangitis, or inflammation of the bile ducts — and the autoimmune mechanism that drives it.
The anti-mitochondrial antibody (AMA) is the serological signature of PBC. The M2 subtype, directed against the E2 subunit of the pyruvate dehydrogenase complex (PDC-E2) on the inner mitochondrial membrane, is detectable in approximately 95% of patients and is highly specific (specificity >95%) for the disease. The 5% of AMA-negative PBC patients typically carry alternative autoantibodies — anti-sp100 or anti-gp210 — that are equally diagnostic in the right clinical context.
PBC sits within the broader spectrum of autoimmune liver diseases alongside autoimmune hepatitis (AIH) and primary sclerosing cholangitis (PSC). An overlap syndrome with AIH occurs in approximately 5-10% of PBC patients and carries a more aggressive course requiring combined immunosuppressive therapy. PBC is also strongly associated with extrahepatic autoimmune conditions: Sjögren's syndrome is present in up to 70% of patients, autoimmune thyroid disease (most commonly Hashimoto's thyroiditis) in approximately 20%, Raynaud's phenomenon in 15-20%, and systemic sclerosis (limited cutaneous type, formerly CREST) in 5-10%.
2. Epidemiology
PBC is relatively uncommon but not rare, with a prevalence ranging from 20 to 40 cases per 100,000 population in Western countries, equating to roughly 100,000 affected individuals in the United States. Incidence estimates range from 0.3 to 5.8 per 100,000 per year depending on geographic region and era of study; rates appear to be increasing over time, at least in part due to improved detection and expanded serological testing.
Geographic clustering is a notable feature: higher rates are consistently reported in Scandinavian countries, the United Kingdom, Canada, and the northern United States compared to Southern Europe, Asia, and the developing world. Within the UK, an unusually high prevalence was documented in northeast England (around Tyneside), which initially suggested possible environmental clustering and spurred early epidemiological investigations.
The typical age of onset is 40 to 60 years, with diagnosis rare before age 25 and uncommon after 70. The striking female predominance (approximately 9 women for every man affected) is one of PBC's defining epidemiological features, though male patients tend to present at more advanced stages and have worse outcomes — possibly due to delayed diagnosis.
Genetic susceptibility plays a meaningful role. First-degree relatives of affected individuals carry a 4-6% lifetime risk of developing PBC, substantially above the general population background. Concordance in identical twins is approximately 60%, confirming a significant genetic contribution while leaving ample room for environmental triggers. Genome-wide association studies have identified multiple susceptibility loci, particularly in HLA class II (HLA-DQB1, HLA-DRB1) and genes involved in T-cell signaling and interleukin pathways (IL-12A, IL-12RB2, STAT4, IRF5).
Environmental triggers remain incompletely understood. Recurrent urinary tract infections (particularly E. coli) have been repeatedly associated with PBC onset, with molecular mimicry between bacterial antigens and PDC-E2 proposed as a mechanism. Xenobiotics — environmental chemicals including halogenated compounds, certain cosmetics (nail polish), and pesticides — have also been implicated, based on case-control studies, but no single environmental trigger has been definitively confirmed.
3. Pathophysiology
PBC results from an autoimmune attack on biliary epithelial cells (cholangiocytes), the specialized cells lining the intrahepatic bile ducts. The primary autoantigen is PDC-E2 (the E2 subunit of the pyruvate dehydrogenase complex), a mitochondrial enzyme involved in aerobic energy metabolism. PDC-E2 is aberrantly expressed on the apical surface of cholangiocytes in PBC patients — a unique vulnerability of this cell type compared to other tissues, which may explain the organ-specific nature of the disease despite the ubiquitous expression of PDC-E2 in all mitochondria-containing cells.
Both humoral and cellular immunity contribute to ductal destruction. The AMA (primarily IgG, IgM subclasses) is a reliable serum marker but is not considered directly pathogenic by itself — AMA-positive individuals who never develop clinical PBC have been documented. The cellular immune attack is believed to be the primary driver: CD4+ helper T cells and CD8+ cytotoxic T cells specific for PDC-E2 infiltrate the portal tracts and launch a sustained attack on cholangiocytes. Molecular mimicry — where immune cells primed against bacterial (particularly E. coli) antigens that share structural homology with PDC-E2 cross-react with bile duct epithelium — is a leading mechanistic hypothesis for how tolerance breaks down.
The consequence of cholangiocyte destruction is impaired bile secretion and bile acid accumulation (cholestasis). Hydrophobic bile acids (deoxycholic acid, lithocholic acid) are directly hepatotoxic and drive secondary hepatocyte injury and inflammation beyond the biliary compartment. Over time, this triggers portal tract fibrosis and the characteristic "biliary-type" fibrosis pattern — fibrous expansion of portal tracts with ductopenia (loss of bile ducts) that distinguishes PBC histologically from other fibrotic liver diseases.
Histological staging by the Ludwig/Scheuer classification defines four stages of increasing severity:
- Stage I (Portal): Florid bile duct lesions — lymphocytic or granulomatous inflammation confined to the portal tracts, often with a characteristic "halo" of lymphocytes surrounding damaged bile ducts.
- Stage II (Periportal): Inflammation spills beyond the portal limiting plate into the periportal parenchyma (interface hepatitis); bile duct proliferation may be seen alongside ongoing duct loss.
- Stage III (Septal/Bridging): Fibrous septa extend between portal tracts (portal-portal bridging fibrosis); progressive ductopenia; inflammatory activity may diminish as ducts are lost.
- Stage IV (Cirrhotic): Regenerative nodules surrounded by fibrous bands; established cirrhosis with its attendant complications of portal hypertension and liver synthetic failure.
It is important to note that histological stages are not always sequential — sampling variability and the inhomogeneous distribution of ductal injury mean that different regions of the same liver may show different stages simultaneously.
4. Clinical Presentation
PBC has a broad and variable clinical spectrum, ranging from asymptomatic biochemical abnormalities detected incidentally on routine blood tests, to advanced symptomatic disease with jaundice, ascites, and liver failure. Increasingly, patients are diagnosed at an asymptomatic stage due to widespread liver function test screening; these patients have a significantly better prognosis than those who present symptomatically.
Fatigue is the most common symptom, affecting 50-78% of PBC patients. It is often profound and disproportionate to the degree of liver damage, significantly impairing quality of life. The mechanism remains poorly understood — it does not correlate with disease severity, ALP levels, or histological stage, and does not reliably improve with UDCA therapy or even liver transplantation in some cases. Hypotheses include autonomic dysfunction, altered central neurotransmitter metabolism, and mitochondrial dysfunction in skeletal muscle.
Pruritus (itching) affects 30-70% of patients at some point and is frequently the presenting symptom. It is caused by the accumulation of bile constituents in the skin — likely lysophosphatidylcholine and endogenous opioids rather than bile salts per se. Pruritus is characteristically worse in the evening and at night, often disrupts sleep, and may be severe enough to cause excoriation and significant psychological distress. It tends to improve in late-stage disease as bile duct destruction becomes complete (less bile production).
Jaundice is uncommon at presentation and signals advanced disease or, when present early, a more aggressive variant or AIH overlap. Its development in a previously compensated PBC patient is a sign of deteriorating hepatic function warranting transplant evaluation.
Additional clinical features include:
- Sicca syndrome (dry eyes and dry mouth): present in up to 70% of patients, usually due to concomitant Sjögren's syndrome.
- Hyperlipidemia: elevated total cholesterol and HDL cholesterol are common in early PBC due to impaired bile acid metabolism and reverse cholesterol transport. Despite elevated total cholesterol, cardiovascular risk does not appear to be significantly elevated — the cholesterol accumulates in HDL particles, which are protective. Xanthelasma (cholesterol deposits around the eyes) and xanthomata (on tendons and pressure points) may develop in patients with severe, prolonged hyperlipidemia.
- Metabolic bone disease: osteoporosis and osteopenia are common complications, driven by impaired absorption of fat-soluble vitamins (particularly vitamin D) due to cholestasis, and by direct effects of bile acids on bone turnover. Bone density is significantly lower than age-matched controls; fracture risk is elevated.
- Raynaud's phenomenon: episodic digital vasospasm in response to cold, present in 15-20% of patients.
- Hypothyroidism: coexists in approximately 20% of PBC patients, most commonly as autoimmune (Hashimoto's) thyroiditis; thyroid-stimulating hormone (TSH) should be checked at diagnosis and periodically thereafter.
- Right upper quadrant discomfort: mild and non-specific; hepatomegaly may be palpable on examination in early disease.
5. Diagnosis and Staging
The diagnosis of PBC is established when at least two of the following three criteria are met:
- Positive AMA: AMA titer ≥1:40 by indirect immunofluorescence, or AMA-M2 positive by ELISA (directed against PDC-E2). AMA is highly sensitive (~95%) and specific (>95%) for PBC.
- Biochemical cholestasis: Elevated alkaline phosphatase (ALP) ≥1.5× the upper limit of normal (ULN), persisting for at least 24 weeks, in the absence of another cause (bone disease, pregnancy, drug effect).
- Compatible liver histology: Non-suppurative destructive cholangitis (florid bile duct lesion) or ductopenia on liver biopsy, consistent with Stage I-IV PBC.
When criteria 1 and 2 are both met, the diagnosis is confirmed without requiring liver biopsy. Biopsy remains indicated to stage disease, assess prognosis, or evaluate for AIH overlap when there is disproportionate transaminase elevation.
Key laboratory findings:
- ALP and GGT: Markedly elevated; these are the hallmark biochemical markers of cholestasis and PBC activity. ALP elevation in isolation (with near-normal AST/ALT) in a middle-aged woman should trigger AMA testing.
- AST and ALT: Usually only modestly elevated (1-3× ULN); marked transaminase elevation (>5× ULN) raises suspicion for AIH overlap.
- Bilirubin: Normal in early disease; rising bilirubin is a significant adverse prognostic marker.
- IgM: Characteristically elevated in PBC; a useful supporting finding.
- ANA patterns: Anti-sp100 (nuclear dot pattern) and anti-gp210 (nuclear rim pattern) are found in approximately 30% of AMA-negative PBC patients and are highly specific; anti-sp100 correlates with more severe disease.
Non-invasive fibrosis assessment: Transient elastography (FibroScan) and serum-based scores (FIB-4, Enhanced Liver Fibrosis [ELF] score) can estimate fibrosis stage and are used to stratify risk and monitor disease progression without repeated biopsies.
UDCA response assessment (Paris criteria): At 12 months of UDCA therapy, biochemical response is assessed using the Paris-II criteria: ALP ≤1.5× ULN AND AST ≤2× ULN AND bilirubin ≤1 mg/dL. Patients meeting these criteria have a near-normal life expectancy; those who fail to respond are at substantially higher risk of disease progression and should be considered for additional therapy.
6. Treatment — UDCA
Ursodeoxycholic acid (UDCA, ursodiol) is the cornerstone first-line treatment for PBC and the only therapy with a decades-long track record of safety and efficacy. The recommended dose is 13-15 mg/kg/day, typically divided into two or three doses and taken with food to maximize absorption and minimize gastrointestinal side effects.
Mechanisms of action: UDCA is a hydrophilic bile acid that works through multiple complementary mechanisms:
- Replaces endogenous toxic hydrophobic bile acids (particularly lithocholic acid and deoxycholic acid) in the bile acid pool, reducing their hepatotoxic effects on cholangiocytes and hepatocytes.
- Stimulates hepatocellular secretion of bile through transcriptional and post-translational upregulation of biliary transporter proteins (BSEP, MDR3, MRP2).
- Exerts immunomodulatory effects, including reduction of aberrant MHC class I expression on cholangiocytes and modulation of cytokine production.
- Reduces apoptosis of cholangiocytes triggered by hydrophobic bile acids.
Clinical outcomes with UDCA:
- Liver biochemistry (ALP, GGT, bilirubin, transaminases) improves significantly in approximately 95% of patients within weeks to months of starting UDCA.
- Prevents or delays histological progression to cirrhosis in patients with early-stage disease (Stages I-II).
- Delays the need for liver transplantation and improves transplant-free survival, particularly in patients who achieve biochemical response.
- Well-tolerated; side effects are uncommon and usually mild (weight gain, hair thinning, diarrhea at higher doses).
Despite these benefits, approximately 40% of patients have an inadequate biochemical response to UDCA as defined by Paris-II criteria at one year. Inadequate responders — particularly those with elevated bilirubin, advanced fibrosis, or younger age — are at significantly higher risk of disease progression and require additional second-line therapy. All patients with PBC, regardless of stage or symptoms, should be offered UDCA unless there is a specific contraindication.
7. Second-Line Therapies
For the approximately 40% of patients with inadequate response to UDCA, several second-line therapies are now available or in active clinical use.
Obeticholic acid (OCA, brand name Ocaliva) was the first approved add-on therapy for PBC with inadequate UDCA response. OCA is a synthetic bile acid analogue that acts as a potent agonist of the farnesoid X receptor (FXR), a nuclear receptor that regulates bile acid synthesis, transport, and metabolism. In the pivotal POISE trial (N Engl J Med 2016), OCA at doses of 5-10 mg/day added to UDCA significantly reduced ALP and bilirubin compared to placebo at 12 months. The recommended starting dose is 5 mg/day, increasing to 10 mg/day after 6 months if tolerated. The major dose-limiting side effect is worsening pruritus, which occurs in a significant proportion of patients and may require dose reduction. OCA is contraindicated in decompensated cirrhosis (Child-Pugh B or C), as cases of hepatic failure and death were reported in this population; it carries a black-box warning accordingly.
Elafibranor (brand name Iqirvo) was approved by the FDA in 2024 for PBC in adults with an inadequate response or intolerance to UDCA. Elafibranor is a dual PPAR-α/δ agonist (peroxisome proliferator-activated receptors alpha and delta) that reduces bile acid synthesis and inflammation. The pivotal ELATIVE trial demonstrated significant reductions in ALP and biochemical response rates versus placebo. Notably, elafibranor does not worsen pruritus — an important advantage over OCA for pruritus-dominant patients.
Seladelpar (brand name Livdelzi), a selective PPAR-δ agonist, was also approved by the FDA in 2024 for PBC with inadequate UDCA response. The RESPONSE trial demonstrated significant ALP reduction and biochemical response, with the additional benefit of improving pruritus scores — making it a particularly attractive option for patients with significant itching.
Management of pruritus: This is one of the most challenging and debilitating symptoms of PBC, often requiring a stepwise approach:
- Cholestyramine (colestipol): Bile acid sequestrant; first-line per major guidelines; must be separated from UDCA by at least 4 hours to prevent binding and reduced absorption of UDCA.
- Rifampicin: Second-line; induces CYP enzymes that metabolize pruritogens; effective in 50-70% of patients; liver function monitoring required (hepatotoxicity risk).
- Naltrexone / naloxone: Opioid antagonists that counteract elevated endogenous opioid tone; moderate evidence of benefit; can trigger opioid withdrawal syndrome at initiation.
- Sertraline: SSRI with modest evidence in PBC-related pruritus; well-tolerated; reasonable option for patients with concurrent mood disturbance.
- Bezafibrate: PPAR agonist fibrate used off-label in Europe; has shown benefit for both pruritus and biochemical response in some trials.
- Plasmapheresis / albumin dialysis (MARS): Reserved for refractory, severe pruritus, particularly as a bridge to transplantation.
Management of fatigue: No pharmacological therapy has demonstrated convincing benefit for PBC-associated fatigue in randomized trials. Regular aerobic exercise and cognitive behavioral therapy show modest benefit. Addressing underlying sleep disturbance (often driven by nocturnal pruritus), depression, anemia, and hypothyroidism is essential before concluding that fatigue is refractory. Fatigue does not reliably improve after liver transplantation in approximately 25-30% of patients, suggesting central nervous system mechanisms rather than purely hepatic ones.
8. Complications
Metabolic bone disease (osteoporosis/osteopenia): Among the most common and clinically significant complications of PBC. Impaired intestinal calcium absorption due to fat-soluble vitamin D deficiency (from cholestasis and reduced fat absorption), direct effects of cholestasis on bone turnover, and potential direct effects of UDCA all contribute. Bone mineral density testing (DEXA scan) should be performed at diagnosis and every 2-3 years thereafter. Management includes:
- Calcium supplementation: 1,000-1,500 mg/day in divided doses
- Vitamin D supplementation: 800-1,000 IU/day (higher doses may be needed if 25-OH vitamin D is low)
- Bisphosphonates (alendronate, risedronate): for established osteoporosis; well-tolerated in PBC; caution with severe esophageal varices
- Weight-bearing exercise to maintain bone density
Fat-soluble vitamin deficiencies (A, D, E, K): Chronic cholestasis impairs micellar solubilization of fat-soluble vitamins, leading to their malabsorption. Vitamin K deficiency may prolong prothrombin time and increase bleeding risk. Vitamins A, D, E, and K levels should be measured at diagnosis and supplemented as needed.
Portal hypertension: May develop in PBC even before cirrhosis is established, through a mechanism called nodular regenerative hyperplasia — sinusoidal remodeling driven by bile acid toxicity that elevates portal pressure without classic fibrotic cirrhosis. Once portal hypertension is established, standard complications of esophageal and gastric varices, ascites, and hepatic encephalopathy can occur and require management per standard protocols.
Hepatocellular carcinoma (HCC): The risk of HCC in PBC is lower than in viral hepatitis-related cirrhosis, but is still significantly elevated compared to the general population, particularly in men and in patients with cirrhosis or inadequate UDCA response. HCC surveillance with liver ultrasound every 6 months is recommended once cirrhosis is established.
PBC-AIH overlap syndrome: Occurs in approximately 5-10% of patients with PBC and is characterized by features of both diseases: AMA positivity alongside prominent interface hepatitis on biopsy, with disproportionately elevated transaminases and IgG levels. This overlap carries a more aggressive course and typically requires combined therapy with UDCA plus corticosteroids (prednisone or budesonide), with azathioprine often added for steroid-sparing maintenance.
Liver transplantation: The definitive treatment for end-stage PBC. Outcomes are excellent, with 5-year post-transplant survival exceeding 80% — among the best of any indication for transplantation. PBC can recur in the transplanted liver in approximately 20-30% of cases, but recurrence is usually mild and rarely leads to graft loss. UDCA is typically continued post-transplant.
9. Prognosis
The prognosis of PBC has improved substantially in the modern treatment era, primarily due to early diagnosis through AMA testing and effective UDCA therapy. For patients who are diagnosed early and respond to UDCA, life expectancy approaches that of the general population.
Several validated prognostic tools are used to stratify risk:
- Mayo PBC Risk Score: Derived from age, serum bilirubin, albumin, prothrombin time, and presence of edema; predicts short- to medium-term survival independent of treatment; used for transplant listing decisions.
- Paris-I and Paris-II Criteria: Biochemical response criteria assessed at 1 year of UDCA therapy; patients meeting these criteria have significantly better transplant-free survival. Paris-II (stricter thresholds) identifies a high-risk group if not met.
- GLOBE Score: An international large-cohort-derived score based on age at treatment initiation, bilirubin, ALP, albumin, and platelet count at 1 year of UDCA; validated to predict 3-, 5-, and 10-year transplant-free survival with high accuracy.
- UK-PBC Risk Score: Combines biochemical parameters at 1 year of UDCA with baseline platelet count and albumin to predict 5-, 10-, and 15-year liver transplant or liver-related death.
Key prognostic determinants include:
- Biochemical response to UDCA: The single strongest modifiable prognostic factor; biochemical responders have near-normal life expectancy.
- Bilirubin level: Rising bilirubin is the most ominous single laboratory marker; patients with bilirubin >2 mg/dL have a median survival of less than 2 years without transplantation.
- Age at diagnosis: Younger patients (under 45) have a higher risk of disease progression despite UDCA — possibly because they have more time for the disease to progress, or because the disease behaves more aggressively in younger individuals.
- Fibrosis stage: Advanced fibrosis (Stage III-IV) at diagnosis confers a worse prognosis regardless of treatment response.
- Male sex: Men tend to present at more advanced stages and have worse outcomes than women, though this may reflect delayed diagnosis rather than inherently different disease biology.
The median time to liver transplantation or death in treated patients diagnosed at an early stage is typically 10-15 years or more from diagnosis. With the newer second-line therapies (OCA, elafibranor, seladelpar) available for UDCA non-responders, the overall trajectory for most patients is increasingly favorable, making PBC one of the more manageable chronic autoimmune liver diseases when identified and treated appropriately.
10. References
- Lindor KD, Bowlus CL, Boyer J, et al. Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019;69(1):394-419. PMID: 30070375
- Nevens F, Andreone P, Mazzella G, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis (POISE). N Engl J Med. 2016;375(7):631-643. PMID: 27532829
- Hirschfield GM, Beuers U, Corpechot C, et al. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67(1):145-172. PMID: 28427765
- Corpechot C, Abenavoli L, Rabahi N, et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology. 2008;48(3):871-877. PMID: 18752324
- Poupon RE, Lindor KD, Cauch-Dudek K, et al. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology. 1997;113(3):884-890. — Search PubMed
- Carbone M, Sharp SJ, Flack S, et al. The UK-PBC risk scores: derivation and validation of a scoring system for long-term prediction of end-stage liver disease in primary biliary cholangitis. Hepatology. 2016;63(3):930-950. PMID: 26223498
- Pares A, Caballeria L, Rodes J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology. 2006;130(3):715-720. PMID: 16530513
- Jones DE, Al-Rifai A, Frith J, et al. The independent effects of fatigue and UDCA therapy on mortality in primary biliary cirrhosis: results of a 9 year follow-up. J Hepatol. 2010;53(5):911-917. — Search PubMed
- Mayo MJ, Parkes J, Adams-Huet B, et al. Prediction of clinical outcomes in primary biliary cirrhosis by serum enhanced liver fibrosis assay. Hepatology. 2008;48(5):1549-1557. — Search PubMed
- Montano-Loza AJ, Carpenter HA, Czaja AJ. Features associated with treatment failure in type 1 autoimmune hepatitis and identification of predictors of treatment response. Hepatology. 2007;45(3):1234-1243. — Search PubMed
- Lammers WJ, van Buuren HR, Hirschfield GM, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology. 2014;147(6):1338-1349. PMID: 25160979
- Trauner M, Nevens F, Shiffman ML, et al. Long-term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: 3-year results of an international open-label extension study. Lancet Gastroenterol Hepatol. 2019;4(6):445-453. — Search PubMed
- Featured Videos
Research Papers
- PBC Diagnosis — PubMed
- UDCA Treatment in PBC — PubMed
- Obeticholic Acid (OCA) in PBC — PubMed
- AMA-M2 Autoantibody — PubMed
- PBC Pruritus Management — PubMed
- PBC Bone Disease — PubMed
- Elafibranor & Seladelpar in PBC — PubMed
- PBC Liver Transplantation — PubMed
- GLOBE Score PBC Prognosis — PubMed
- PBC-AIH Overlap Syndrome — PubMed
- PBC Epidemiology — PubMed
- PDC-E2 Autoantigen Pathophysiology — PubMed
Connections
- Nephrology & Hepatology
- Cirrhosis
- Liver Disease
- Non-Alcoholic Fatty Liver Disease
- Hepatic Encephalopathy
- Gallbladder Disease
- Autoimmune Disease
- Hypothyroidism
- Vitamin D
- Vitamin K
- Osteoporosis
- GGT
- Alkaline Phosphatase (ALP)