Vitamin B6 for Neurotransmitter Synthesis (Serotonin, Dopamine, GABA)
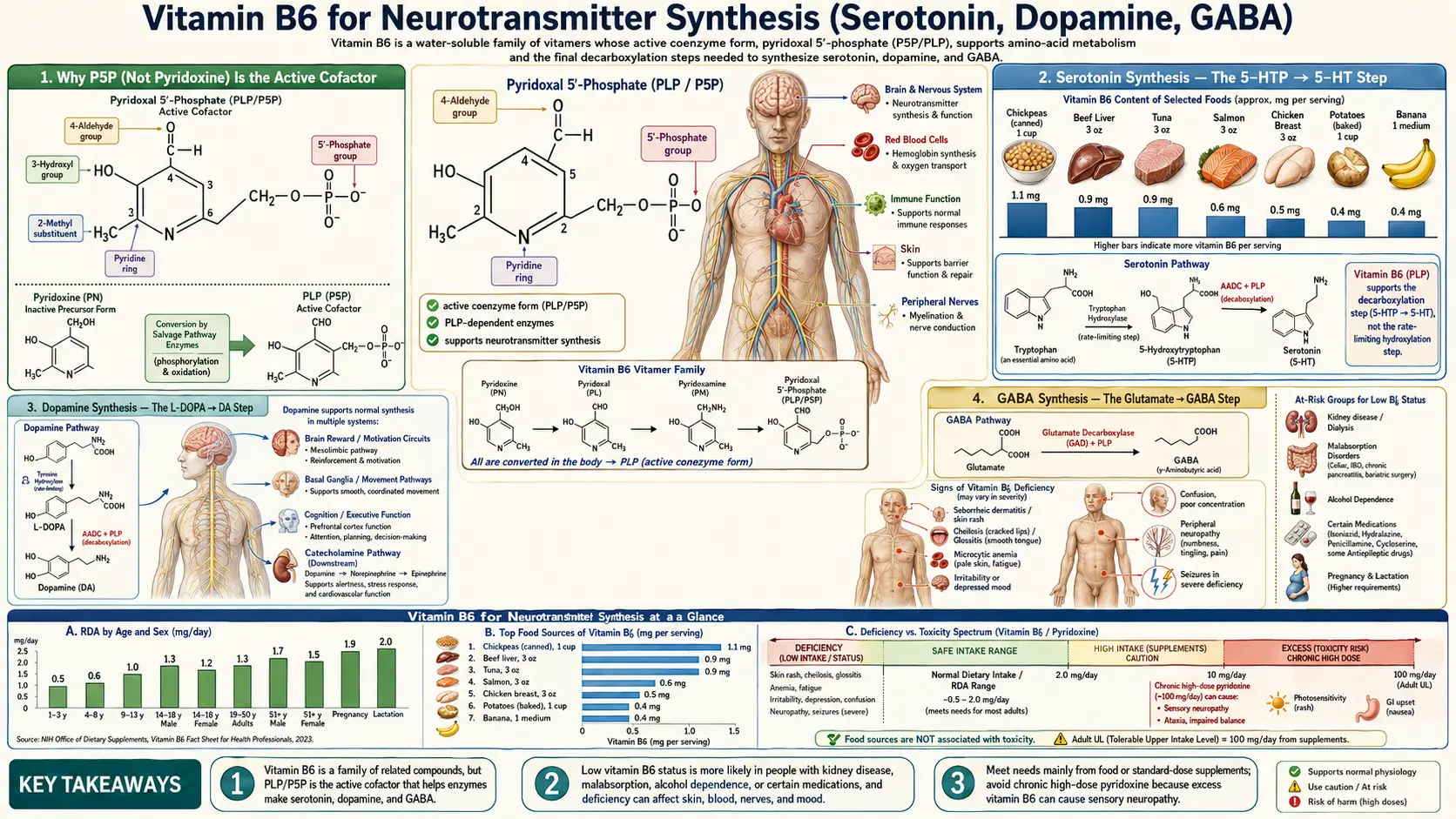
Pyridoxal-5-phosphate (P5P), the active coenzyme form of Vitamin B6, is the obligate cofactor for the enzymes that synthesize serotonin, dopamine, norepinephrine, GABA, and histamine. Without adequate P5P, the brain cannot manufacture its primary excitatory and inhibitory neurotransmitters. The clinical consequences range from depression and anxiety in modest deficiency to outright seizures (pyridoxine-dependent epilepsy) in severe deficiency. This deep dive covers the biochemistry, the clinical evidence in mood disorders, the autism+B6/magnesium protocols, the rare but treatable PDE/PNPO genetic epilepsies, and where B6 fits within an integrative neuropsychiatric protocol.
Table of Contents
- Why P5P (Not Pyridoxine) Is the Active Cofactor
- Serotonin Synthesis — The 5-HTP → 5-HT Step
- Dopamine Synthesis — The L-DOPA → DA Step
- GABA Synthesis — The Glutamate → GABA Step
- Norepinephrine, Epinephrine, and Histamine
- Why One Enzyme (AADC) Makes Both Serotonin and Dopamine
- Why B6 Deficiency Causes Seizures
- Pyridoxine-Dependent Epilepsy (PDE, ALDH7A1)
- PNPO Deficiency and P5P-Responsive Epilepsy
- Autism Spectrum — The B6 + Magnesium Protocols
- Depression and Anxiety Augmentation
- SSRI Synergy and Practical Considerations
- Practical Dosing Protocol
- Cautions
- Key Research Papers
- Connections
- Featured Videos
Why P5P (Not Pyridoxine) Is the Active Cofactor
Of the three Vitamin B6 forms (pyridoxine, pyridoxal, pyridoxamine), only pyridoxal-5-phosphate (P5P) is enzymatically active. P5P participates in catalysis by forming a Schiff base (covalent imine bond) with the alpha-amino group of an amino acid substrate. The aromatic pyridine ring then stabilizes the resulting carbanion intermediate — this is the chemical magic that allows P5P-dependent enzymes to perform transamination, decarboxylation, racemization, and beta-elimination on essentially any amino acid.
For supplemental pyridoxine to support neurotransmitter synthesis, it must be activated in two steps: first phosphorylation by pyridoxal kinase (requires zinc + magnesium + ATP), then oxidation by pyridoxine phosphate oxidase (PNPO) — an FMN-dependent enzyme that requires adequate riboflavin (B2). In people with riboflavin deficiency, PNPO polymorphisms, hepatic dysfunction, or rare PNPO mutations, this activation step fails and supplemental pyridoxine cannot reach the brain in a useful form. Direct P5P supplementation bypasses both steps and is the preferred form for neuropsychiatric applications.
Serotonin Synthesis — The 5-HTP → 5-HT Step
Serotonin (5-hydroxytryptamine, 5-HT) is synthesized in two enzymatic steps from dietary tryptophan:
- Tryptophan + O&sub2; → 5-hydroxytryptophan (5-HTP), catalyzed by tryptophan hydroxylase (TPH1/TPH2). This is the rate-limiting step and requires tetrahydrobiopterin (BH4), iron, and molecular oxygen.
- 5-HTP → serotonin, catalyzed by aromatic L-amino acid decarboxylase (AADC) — a P5P-dependent enzyme. This is the B6 step.
Without P5P, the brain accumulates 5-HTP and cannot make serotonin. The clinical consequences are predictable: depressed mood, anxiety, irritability, insomnia (because serotonin is the precursor to melatonin), carbohydrate cravings, and reduced pain tolerance.
This is why tryptophan or 5-HTP supplementation alone is often disappointing for depression: it adds substrate to a pathway that may already be cofactor-limited. The integrative-medicine combination of 5-HTP + P5P (typically 100 mg 5-HTP + 25-50 mg P5P) addresses both substrate and cofactor limitations simultaneously.
Dopamine Synthesis — The L-DOPA → DA Step
Dopamine synthesis follows a structurally identical pathway from tyrosine (or phenylalanine → tyrosine):
- Tyrosine + O&sub2; → L-DOPA (3,4-dihydroxyphenylalanine), catalyzed by tyrosine hydroxylase. Rate-limiting; requires BH4, iron, oxygen.
- L-DOPA → dopamine, catalyzed by aromatic L-amino acid decarboxylase (AADC) — the same P5P-dependent enzyme that decarboxylates 5-HTP. This is the B6 step.
The clinical consequences of impaired dopamine synthesis include: low motivation/anhedonia, poor focus and working memory, reduced motor activation, increased prolactin (because dopamine normally suppresses prolactin via the tubero-infundibular pathway), and contribution to depressive symptoms with a "low-energy" rather than "anxious-agitated" phenotype.
The pharmaceutical Parkinson's disease combination drug Sinemet exploits this enzymatic identity: levodopa is paired with carbidopa, a peripheral AADC inhibitor that prevents premature decarboxylation in the gut and liver. Pyridoxine in modest amounts is required for the central conversion of L-DOPA to dopamine inside the brain. Historically, high-dose pyridoxine was warned against in Parkinson's patients on levodopa monotherapy because it accelerated peripheral conversion, but with the carbidopa combination this is no longer clinically relevant at normal B6 intakes.
GABA Synthesis — The Glutamate → GABA Step
GABA (gamma-aminobutyric acid) is the brain's primary inhibitory neurotransmitter, accounting for the calming activity of benzodiazepines, gabapentin, alcohol, and barbiturates. GABA is synthesized in a single enzymatic step:
- Glutamate → GABA + CO&sub2;, catalyzed by glutamate decarboxylase (GAD). GAD is a P5P-dependent enzyme. Two isoforms exist: GAD65 (located in synaptic terminals, synthesizes the rapidly-mobilized pool of GABA) and GAD67 (cytoplasmic, synthesizes the bulk constitutive pool).
This single reaction has outsized clinical importance for two reasons. First, GAD converts the brain's most abundant excitatory neurotransmitter (glutamate) into its most abundant inhibitory neurotransmitter (GABA). The same molecule of glutamate goes from "exciting neurons" to "inhibiting neurons" in one P5P-dependent step — B6 status literally flips the brain's excitatory/inhibitory balance.
Second, GAD is the slowest-saturating P5P-dependent enzyme in the brain. When P5P levels fall (deficiency, alcohol use, inflammation, isoniazid, hydrazine exposure), GAD activity collapses first while other P5P enzymes still function. The result is a relative excess of glutamate combined with a deficit of GABA — the biochemical recipe for seizures, anxiety, insomnia, and irritability.
Norepinephrine, Epinephrine, and Histamine
P5P participates in or supports the synthesis of several additional neuroactive amines:
- Norepinephrine — produced from dopamine by dopamine beta-hydroxylase (requires copper and vitamin C, not P5P). However, because dopamine itself requires P5P-dependent AADC, B6 deficiency reduces norepinephrine indirectly through reduced substrate availability.
- Epinephrine (adrenaline) — produced from norepinephrine by phenylethanolamine N-methyltransferase (requires SAMe, indirectly B6/B12/folate via the methylation cycle).
- Histamine — produced from histidine by histidine decarboxylase, a P5P-dependent enzyme. While histamine is best known as an allergic mediator, it also functions as a neurotransmitter regulating wakefulness, gastric acid secretion, and certain cognitive functions. B6-deficient patients sometimes paradoxically present with reduced histamine signaling and excessive sleepiness, fatigue, or hypochlorhydria.
- Melatonin — produced from serotonin by N-acetylserotonin via the methylation cycle. Because B6 is required for serotonin synthesis, it indirectly supports nighttime melatonin production and healthy circadian rhythm.
- Taurine — not a classical neurotransmitter but a major brain inhibitory amino acid that supports GABA-A receptor function. Taurine synthesis from cysteine requires P5P-dependent cysteine sulfinic acid decarboxylase.
Why One Enzyme (AADC) Makes Both Serotonin and Dopamine
An elegant economy of evolution: a single P5P-dependent enzyme — aromatic L-amino acid decarboxylase — performs the final synthetic step for both serotonin (5-HTP → 5-HT) and dopamine (L-DOPA → DA). This is because both substrates share the same key chemical feature: an aromatic amino acid with a free alpha-amino group. P5P's Schiff-base mechanism is indifferent to whether the side chain is the indole of 5-HTP or the catechol of L-DOPA.
The clinical implication is that B6 deficiency tends to depress serotonin AND dopamine in parallel, producing a "dual depletion" phenotype that combines anxious-irritable features (low serotonin) with low-motivation/anhedonic features (low dopamine). Patients respond particularly poorly to single-target SSRI therapy because the unaddressed dopamine deficit limits clinical improvement.
Rare AADC deficiency (a genetic disease, biallelic DDC mutations) presents in infancy with profound motor delay, autonomic dysregulation, oculogyric crises, and developmental delay. Treatment is partially responsive to high-dose pyridoxine (which can drive residual AADC enzyme activity through cofactor saturation), dopamine agonists, MAO inhibitors, and now (since 2022) the gene therapy eladocagene exuparvovec approved in the EU.
Why B6 Deficiency Causes Seizures
Severe Vitamin B6 deficiency — whether from dietary lack, isoniazid (the tuberculosis drug, which directly inhibits pyridoxal kinase and complexes with P5P), gyromitrin mushroom poisoning, hydrazine exposure, or genetic disorders — reliably causes convulsive seizures. The mechanism is the GAD/GABA collapse described above.
When P5P drops below a critical threshold, glutamate decarboxylase loses its cofactor. GABA synthesis stops. Glutamate accumulates. The excitatory/inhibitory balance shifts so dramatically toward excitation that the cortex spontaneously hyperexcites — a classic generalized seizure.
In infants and adults presenting with status epilepticus refractory to standard antiseizure medications, the empirical IV pyridoxine challenge (100 mg IV in adults, 50-100 mg IV in neonates) is a standard ER protocol. If the seizure breaks within minutes, pyridoxine-responsive epilepsy is the diagnosis. This is the rationale behind the "100 mg IV pyridoxine and 100 mg thiamine" combination given empirically in unexplained adult seizures or coma.
Pyridoxine-Dependent Epilepsy (PDE, ALDH7A1)
Pyridoxine-Dependent Epilepsy is a rare autosomal recessive metabolic disorder caused by mutations in ALDH7A1 (encoding antiquitin, an aldehyde dehydrogenase). Affected children typically present with neonatal-onset, refractory seizures within the first hours to weeks of life.
The mechanism: ALDH7A1 normally catalyzes a step in lysine degradation. When ALDH7A1 fails, an upstream metabolite called alpha-aminoadipic semialdehyde (alpha-AASA) accumulates. Alpha-AASA chemically reacts with P5P, depleting it from the brain. The result is exactly the GAD/GABA collapse described above — severe inherited functional B6 deficiency despite normal dietary intake.
Treatment: Pharmacological doses of pyridoxine (typically 30–100 mg/kg/day in infants, 100–500 mg/day in older children and adults) saturate the system and overcome the P5P depletion. Lysine-restricted diet plus arginine supplementation has been added in modern protocols to reduce alpha-AASA accumulation at its source. Lifelong treatment is required; missed doses produce immediate seizure recurrence.
The key clinical pearl: PDE seizures are refractory to phenobarbital, phenytoin, levetiracetam, and all standard antiseizure drugs, but respond dramatically to pyridoxine within minutes to hours. Any infant with refractory neonatal seizures should receive a pyridoxine trial.
PNPO Deficiency and P5P-Responsive Epilepsy
A closely related but distinct disorder: pyridox(am)ine 5'-phosphate oxidase (PNPO) deficiency. Recall that PNPO is the enzyme that converts pyridoxine phosphate or pyridoxamine phosphate into the active P5P. In children with biallelic PNPO mutations, supplemental pyridoxine cannot be activated to P5P and is therefore therapeutically useless.
These children present with neonatal seizures that are refractory to standard antiseizure drugs AND to pyridoxine, but respond dramatically to pyridoxal-5-phosphate (P5P) directly. Typical dose: 30–50 mg/kg/day P5P, divided. Lifelong supplementation is required.
This is the strongest single argument for keeping P5P (not just pyridoxine HCl) in clinical pharmacopoeias and emergency departments worldwide. The clinical decision tree in neonatal refractory seizures should be: (1) standard antiseizure drugs → if refractory, (2) IV pyridoxine challenge → if refractory, (3) IV P5P challenge.
Autism Spectrum — The B6 + Magnesium Protocols
Bernard Rimland (founder of the Autism Research Institute) published the original B6+magnesium trials for autism in the 1970s. Across approximately 18 small studies in the following decades, a subgroup of children with autism spectrum disorder showed clinically meaningful improvement on combined high-dose pyridoxine (typically 8–16 mg/kg/day, often 200–500 mg/day in older children and adolescents) plus magnesium (typically 4–8 mg/kg/day, often 200–400 mg/day).
The proposed mechanism: a subgroup of autistic children have evidence of impaired P5P-dependent neurotransmitter synthesis (reduced urinary metabolites of serotonin and dopamine; reduced CSF concentrations of these neurotransmitters). The B6+magnesium combination addresses both the cofactor (P5P) and a critical secondary cofactor (magnesium, required for pyridoxal kinase activation of supplemental pyridoxine).
Clinical reality: the response rate in published trials is approximately 30-50% of trialed children — not all autistic children respond, but a substantial minority show measurable improvement in eye contact, language, behavior, or repetitive movements over 4-8 weeks. The 2005 Cochrane review concluded the evidence was inadequate to recommend the combination as standard practice, but many integrative pediatricians continue to offer time-limited trials of B6+magnesium in autistic children with appropriate monitoring.
The toxicity concern is real and must be respected. Doses of 200–500 mg/day pyridoxine are exactly in the range associated with adult sensory neuropathy. P5P (rather than pyridoxine HCl) should be used. Maximum trial duration should be 8-12 weeks before assessing response, and benefit must be documented to justify continuation. Children should have neurological exams (especially gait, balance, and deep tendon reflexes) before starting and every 3 months thereafter. See the Toxicity deep dive.
Depression and Anxiety Augmentation
B6 status correlates inversely with depressive symptoms in cross-sectional studies. Low plasma P5P is found consistently in patients with major depression, and the deficit often persists despite SSRI or SNRI treatment. The mechanism is straightforward — serotonin synthesis is rate-limited by P5P-dependent AADC activity.
For anxiety, a 2022 randomized controlled trial by Field et al. (University of Reading) tested 100 mg pyridoxine HCl daily for one month versus placebo in 478 young adults. The B6 group showed statistically significant reductions in anxiety scores compared to placebo, with the effect attributable to GAD/GABA enhancement. The trial duration was too short to raise toxicity concerns but the dose (100 mg) is at the upper safe limit and should not be continued indefinitely without monitoring.
For depression, no large randomized monotherapy trials exist, but small studies of P5P augmentation of SSRI therapy (typically 25-50 mg P5P added to a stable SSRI dose) have shown improved response rates and faster onset of effect. This is a common integrative-psychiatry practice but lacks definitive RCT evidence.
For premenstrual dysphoric disorder (PMDD), the Wyatt meta-analysis (covered in the PMS deep dive) found moderate-strength evidence for 50–100 mg pyridoxine daily, with NNT approximately 3 for mood symptoms and NNT approximately 9 for depression specifically.
SSRI Synergy and Practical Considerations
SSRIs (sertraline, escitalopram, fluoxetine, etc.) work by blocking serotonin reuptake, leaving more 5-HT in the synapse to act on receptors. They do not increase serotonin synthesis. In a patient with B6 deficiency, the synthesis bottleneck remains unaddressed — the SSRI can only redistribute the inadequate serotonin pool, producing partial or absent clinical response.
The integrative-psychiatry observation is that SSRI non-responders often have low plasma P5P. Adding 25–50 mg P5P to a stable SSRI regimen frequently produces additional clinical improvement that the SSRI alone could not deliver. This is anecdotal but biologically plausible and low-risk at these doses.
SNRIs (venlafaxine, duloxetine) face the same constraint, with the additional dopamine/norepinephrine bottleneck through the same AADC enzyme. P5P augmentation can theoretically improve both serotonergic and noradrenergic activity simultaneously.
Tricyclic antidepressants, MAOIs, and atypical antidepressants (bupropion, mirtazapine) all share the dependence on adequately synthesized monoamines. Pyridoxine/P5P adequacy is a precondition for all of them to work optimally.
Practical Dosing Protocol
For mood/anxiety augmentation in healthy adults
- P5P 25–50 mg/day with breakfast (energizing; avoid at bedtime)
- Take as part of a B-complex containing riboflavin (B2), folate, and B12 — isolated B6 supplementation can deplete the others over time
- Pair with magnesium glycinate 200–400 mg/day — magnesium is required for pyridoxal kinase
- Trial duration: 4–8 weeks to assess effect
- Maximum sustained dose: 100 mg/day total B6 (FDA Upper Limit). Do not exceed without specific indication and monitoring.
For SSRI/SNRI augmentation
- Same as above; coordinate with prescribing physician
- P5P-form preferred over pyridoxine HCl for any chronic use at 50 mg+
For pyridoxine-dependent epilepsy or PNPO deficiency
- Specialist (pediatric neurology) management only
- PDE: pyridoxine 30–100 mg/kg/day in infants; 100–500 mg/day in older children/adults
- PNPO: P5P specifically required at 30–50 mg/kg/day, divided
- Lifelong therapy; missed doses produce seizure recurrence
- Lysine-restricted diet + arginine supplementation per modern PDE protocols
For autism B6+magnesium trial
- Specialist (integrative pediatrics) supervision strongly recommended
- P5P (not pyridoxine HCl) preferred
- Trial 8–12 weeks; document response before continuing
- Neurological exam (gait, balance, deep tendon reflexes) baseline and every 3 months
- Discontinue at first sign of paresthesias or gait instability
Cautions
- Sensory neuropathy is the dose-limiting toxicity — chronic doses above 100 mg/day pyridoxine HCl, and possibly above 200 mg/day of P5P, can cause peripheral sensory neuropathy. See the Toxicity deep dive for full discussion. Use the minimum effective dose; trial at higher doses for the shortest period needed; switch to P5P form when possible.
- Isoniazid interaction — the tuberculosis drug isoniazid directly antagonizes P5P (forms a hydrazone, inhibits pyridoxal kinase). All patients on isoniazid should receive 25–50 mg pyridoxine daily to prevent neuropathy. This is standard TB clinic practice.
- Levodopa interaction (legacy concern) — high-dose pyridoxine (>200 mg/day) historically accelerated peripheral L-DOPA conversion before patients reached the brain, reducing efficacy of unprotected levodopa. With modern carbidopa/levodopa combinations, normal B6 intakes are no longer problematic.
- Phenytoin and phenobarbital interaction — chronic anticonvulsant use can deplete B6 and cause functional deficiency; supplementation may be needed but should be modest (25 mg/day) to avoid altering antiseizure drug pharmacokinetics.
- Penicillamine, hydralazine, cycloserine — all are B6 antagonists; supplementation indicated.
- Oral contraceptives — significantly deplete B6; supplement 25–50 mg/day routinely. See the PMS & Hormonal deep dive.
- Pregnancy — pyridoxine 10–75 mg/day is well-established as safe and effective for nausea/vomiting of pregnancy and is a Category A drug. See the PMS & Hormonal deep dive.
Key Research Papers
- Hatch J et al. (2013). Aromatic L-amino acid decarboxylase deficiency: review of treatment outcomes. Journal of Inherited Metabolic Disease. — PubMed
- Mills PB et al. (2014). Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nature Medicine. — PubMed
- Mills PB et al. (2005). Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5'-phosphate oxidase. Human Molecular Genetics. — PubMed
- Stockler S et al. (2011). Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Molecular Genetics and Metabolism. — PubMed
- Field DT et al. (2022). High-dose Vitamin B6 supplementation reduces anxiety and strengthens visual surround suppression. Human Psychopharmacology. — PubMed
- Adams JB et al. (2011). Effect of a vitamin/mineral supplement on children and adults with autism. BMC Pediatrics. — PubMed
- Nye C, Brice A (2005). Combined vitamin B6-magnesium treatment in autism spectrum disorder. Cochrane Database. — PubMed
- Rimland B, Callaway E, Dreyfus P (1978). The effect of high doses of vitamin B6 on autistic children: a double-blind crossover study. American Journal of Psychiatry. — PubMed
- Williams AL et al. (2005). Vitamin B6 augmentation of antidepressants in depression. Nutrition Reviews. — PubMed
- Hellmann H, Mooney S (2010). Vitamin B6: a molecule for human health? Molecules. — PubMed
- Coursin DB (1954). Convulsive seizures in infants with pyridoxine-deficient diet. JAMA. The original report. — PubMed
PubMed Topic Searches
- PubMed: B6 neurotransmitter synthesis
- PubMed: pyridoxine-dependent epilepsy
- PubMed: PNPO deficiency
- PubMed: B6 + magnesium autism
- PubMed: GAD P5P seizures
Connections
- Vitamin B6 Overview
- B6 Benefits Hub
- B6 for PMS & Hormonal
- B6 for Homocysteine
- B6 Toxicity (READ FIRST)
- Pyridoxine and Brain Health
- Vitamin B2 (Riboflavin — required for B6 activation)
- Vitamin B9 (Folate)
- Vitamin B12
- Magnesium
- Tryptophan
- Glycine
- Depression
- Anxiety
- Insomnia
- Peripheral Neuropathy