Niacin for Cholesterol and Cardiovascular Disease
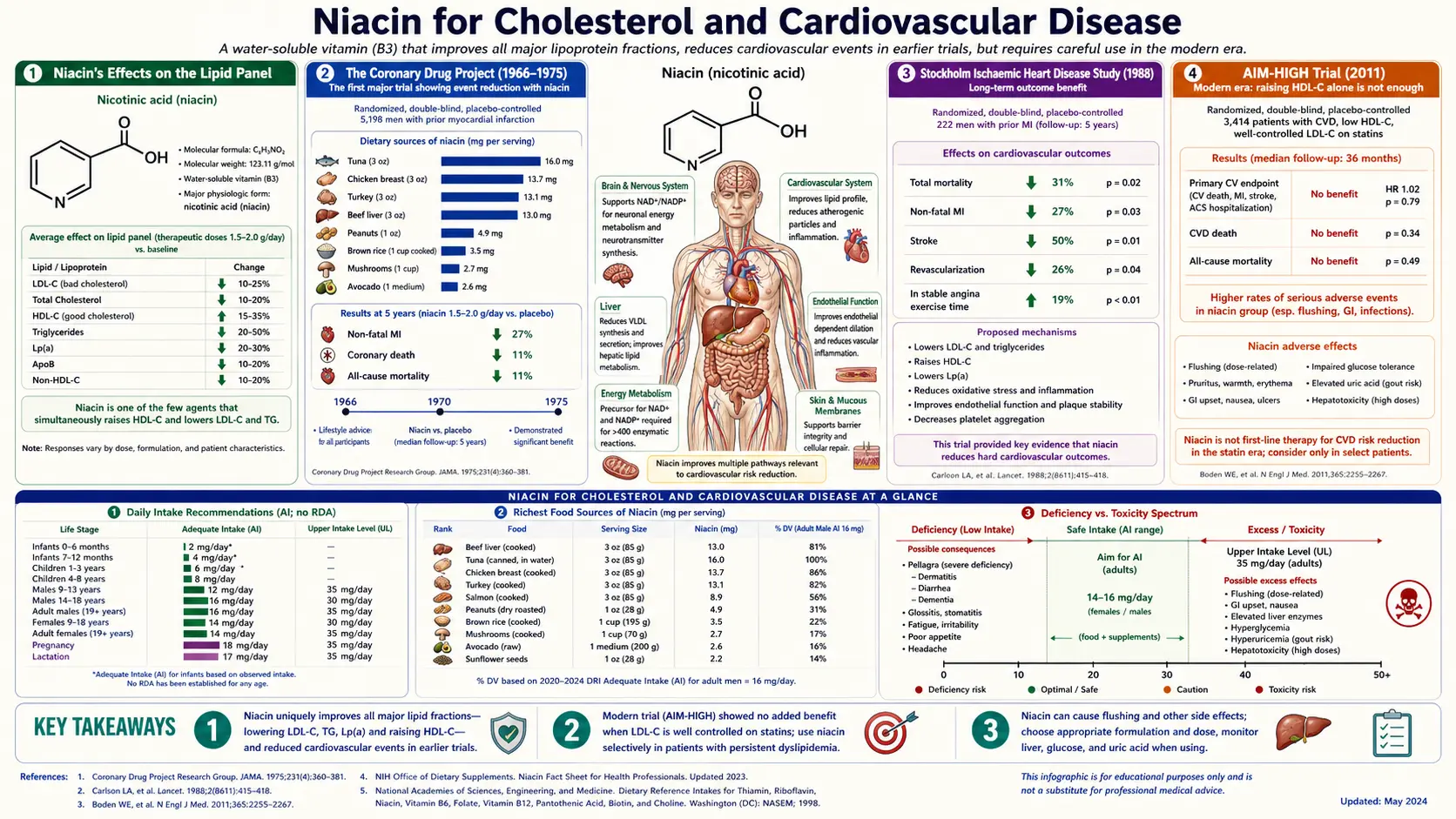
Nicotinic acid (immediate-release niacin) was the first lipid-modifying drug ever shown to reduce all-cause mortality in a randomized trial — the 1975 Coronary Drug Project, with the benefit persisting at the 15-year follow-up published by Canner in 1986. It remains the most potent HDL-raising agent ever discovered and one of the only readily available drugs that meaningfully lowers lipoprotein(a). Two large negative trials in the 2010s — AIM-HIGH and HPS2-THRIVE — correctly led mainstream cardiology to abandon niacin as an add-on to optimized statin therapy, but they did not invalidate its role as monotherapy in statin-intolerant patients or as a targeted intervention for elevated Lp(a) and ApoB. This page walks the trial history honestly and gives the modern 1-3 g/day immediate-release titration protocol that still has a place in 2026 practice.
Table of Contents
- Niacin's Effects on the Lipid Panel
- The Coronary Drug Project (1966-1975)
- Stockholm Ischaemic Heart Disease Study (1988)
- AIM-HIGH (2011)
- HPS2-THRIVE (2014)
- Why the Modern Trials Were Negative
- Modern Positioning — When Niacin Still Makes Sense
- The Lipoprotein(a) Question
- Mechanism of Lipid Modification
- 1-3 g/day Immediate-Release Titration Protocol
- Cautions
- Key Research Papers
- Connections
- Featured Videos
Niacin's Effects on the Lipid Panel
At pharmacological doses of immediate-release nicotinic acid (1000-3000 mg/day), no single agent matches niacin's spectrum of lipid effects:
| Lipid Marker | Niacin Effect (1-3 g/day IR) | Comparable Agents |
|---|---|---|
| HDL cholesterol | +15 to +35% | No drug matches this |
| LDL cholesterol | −5 to −25% | Statins (much stronger); ezetimibe; PCSK9 inhibitors |
| Triglycerides | −20 to −50% | Fibrates; high-dose omega-3 (icosapent ethyl) |
| Lipoprotein(a) [Lp(a)] | −20 to −30% | PCSK9 inhibitors (similar); pelacarsen (investigational); apheresis |
| ApoB | −15 to −25% | Statins; ezetimibe; PCSK9 inhibitors |
| LDL particle pattern | Shifts small-dense to large-buoyant (less atherogenic) | Fibrates (similar) |
The HDL-raising and Lp(a)-lowering effects are essentially unique to niacin among broadly available drugs. PCSK9 inhibitors (alirocumab, evolocumab) lower Lp(a) by similar magnitudes but cost $5000-7000/year — thousands of times more than niacin. The Lp(a) effect, in particular, makes niacin clinically interesting in 2026 because elevated Lp(a) is an underrecognized cardiovascular risk factor with limited therapeutic options.
For the broader picture of niacin's lipid effects in our existing literature, see Niacin & Cholesterol (the long-form article) and Cholesterol Management.
The Coronary Drug Project (1966-1975)
The Coronary Drug Project (CDP) was a landmark National Heart, Lung, and Blood Institute trial of secondary prevention in 8341 men aged 30-64 who had survived at least one myocardial infarction. Participants were randomized to one of five lipid-modifying interventions or placebo and followed for 5-8.5 years.
The five treatment arms
- Niacin (immediate-release nicotinic acid, 3000 mg/day) — 1119 patients
- Clofibrate (1800 mg/day)
- Two estrogen doses (both discontinued early for safety)
- Dextrothyroxine (discontinued early for cardiovascular harm)
- Placebo — 2789 patients
The CDP 1975 publication (JAMA)
- Niacin was the only lipid-modifying treatment to show benefit.
- 14% reduction in nonfatal recurrent MI (p < 0.005)
- 27% reduction in cerebrovascular events (TIA + stroke)
- Total mortality during the active treatment period showed a trend toward benefit but did not reach significance at the 5-8 year endpoint
- Clofibrate showed no significant benefit on any cardiovascular endpoint
The Canner 1986 follow-up (JACC) — the mortality benefit emerges
Paul Canner reanalyzed the CDP cohort 15 years after the trial began — 9 years after active treatment had ended. The result is one of the most striking long-term follow-up findings in cardiovascular medicine:
- 11% reduction in all-cause mortality in the niacin arm versus placebo at 15 years (p = 0.0004)
- The benefit had emerged after active treatment ended — a "legacy effect" similar to what was later seen with statins in the UKPDS follow-up
- The mortality benefit was driven primarily by reduced cardiovascular deaths but also included reduced non-cardiovascular mortality
- This made nicotinic acid the first lipid-modifying drug ever shown to reduce all-cause mortality in a randomized controlled trial
The CDP/Canner result remained the dominant cardiovascular evidence for niacin for the next 25 years, supporting widespread clinical use of niacin (often combined with bile-acid sequestrants and later with low-dose statins) for cardiovascular prevention.
Stockholm Ischaemic Heart Disease Study (1988)
Lars Carlson and Goran Rosenhamer published the Stockholm Ischaemic Heart Disease Secondary Prevention Study in Acta Medica Scandinavica in 1988. 555 post-MI patients were randomized to standard care or to combination nicotinic acid (3000 mg/day) plus clofibrate (2000 mg/day), and followed for 5 years.
- 26% reduction in all-cause mortality (p < 0.05)
- 36% reduction in ischemic heart disease mortality
- The benefit was largest in patients with the greatest triglyceride reduction
The Stockholm study was a second positive RCT in the pre-statin era, reinforcing the CDP findings. Notably, both the CDP and the Stockholm trials used immediate-release nicotinic acid at gram-range doses — not extended-release formulations.
AIM-HIGH (2011)
AIM-HIGH (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes) randomized 3414 patients with established atherosclerotic cardiovascular disease and atherogenic dyslipidemia (low HDL, high triglycerides) to:
- Extended-release niacin (Niaspan) 1500-2000 mg/day plus simvastatin (with ezetimibe added as needed) targeting LDL < 80 mg/dL
- OR placebo plus the same statin regimen
The trial was stopped early at 3 years for futility. Published in NEJM 2011:
- No reduction in the primary composite endpoint (CHD death, nonfatal MI, ischemic stroke, hospitalization for ACS, or symptom-driven coronary/cerebral revascularization)
- Niacin successfully raised HDL by 25% and lowered triglycerides by 29%, but this lipid improvement did not translate into clinical events benefit
- A non-significant numerical excess of ischemic stroke in the niacin arm
The trial had design limitations — small placebo dose of niacin (50 mg) was given to mask the flush, which may have created lipid effects in the "placebo" group; the population had already achieved aggressive LDL lowering on statin before randomization, leaving little room for incremental benefit; and the trial was underpowered after early stopping.
HPS2-THRIVE (2014)
HPS2-THRIVE (Heart Protection Study 2 — Treatment of HDL to Reduce the Incidence of Vascular Events) was the larger trial that effectively ended mainstream cardiology's use of niacin. 25,673 patients with vascular disease already taking effective LDL-lowering therapy (mostly simvastatin 40 mg) were randomized to:
- Extended-release niacin (Niaspan) 2000 mg/day plus laropiprant 40 mg/day (a prostaglandin D2 receptor antagonist intended to suppress flushing)
- OR placebo
Median follow-up was 3.9 years. Published in NEJM 2014:
- No reduction in major vascular events
- Significant increase in serious adverse events: new-onset diabetes (+1.8% absolute), diabetic complications, GI events, musculoskeletal events, skin events, and infections
- 4% absolute increase in disturbances of diabetes control among diabetic patients
- Increased gout, dyspepsia, diarrhea, and rash
HPS2-THRIVE was definitive that extended-release niacin plus laropiprant adds harm without benefit when given on top of optimized statin therapy. Merck withdrew the niacin/laropiprant combination (Tredaptive/Cordaptive) from market globally. The FDA pulled approval for niacin add-on to statins (Advicor and Simcor combination products were withdrawn in 2016).
Why the Modern Trials Were Negative
The collapse of niacin as add-on to statins did not invalidate the earlier evidence. Several factors explain the divergence:
- Background statin therapy was the difference. CDP (1975) was conducted before statins existed — placebo patients had LDL of ~130-140 mg/dL on average. AIM-HIGH and HPS2-THRIVE patients had LDL already aggressively lowered to ~70 mg/dL by statin therapy. The marginal benefit of additional HDL-raising or further LDL-lowering at these already-low levels appears minimal.
- The "HDL hypothesis" itself was wrong. Mendelian randomization studies (CETP variants and others) have shown that genetically determined HDL levels do not causally protect against CHD. HDL is a marker of cardiometabolic health, not a causal protective particle. Drugs that raise HDL (CETP inhibitors like dalcetrapib, evacetrapib, anacetrapib; niacin) have failed to translate HDL gains into outcomes.
- Laropiprant added cardiovascular harm. The flush-blocking adjunct used in HPS2-THRIVE was implicated in some of the excess adverse events — HPS2-THRIVE was not a pure test of niacin alone.
- Extended-release vs immediate-release. Both modern trials used ER-niacin. The historical positive trials (CDP, Stockholm) used IR-niacin at higher peak concentrations. Whether IR vs ER pharmacokinetics matter for outcomes is an unresolved question.
- LDL particle composition matters more than LDL concentration. Niacin's benefit may operate primarily through ApoB and Lp(a) reduction and LDL-particle-size shift, which are not directly captured by standard lipid panels.
The honest reading of the totality of evidence: niacin works when it is the primary lipid-modifying agent in patients who would otherwise have untreated dyslipidemia; it does not add incremental benefit when stacked on top of optimized statin therapy.
Modern Positioning — When Niacin Still Makes Sense
In 2026 cardiology practice, niacin's legitimate clinical niches are narrower than they were in 1995 but remain real:
- Statin intolerance. Patients with statin-associated muscle symptoms, statin-induced liver enzyme elevation, or PCSK9-inhibitor cost barriers may need an alternative lipid-modifying agent. Immediate-release niacin at 1-3 g/day is a reasonable monotherapy choice for LDL reduction and HDL raising, with the caveat that LDL reduction is much weaker than with statins.
- Elevated Lp(a). Lp(a) above 50 mg/dL (or 125 nmol/L) is an independent cardiovascular risk factor with limited therapeutic options. Niacin at 1-2 g/day reduces Lp(a) by 20-30%. Until pelacarsen and other antisense Lp(a) drugs are available, niacin remains one of the most cost-effective Lp(a)-lowering options.
- Severe hypertriglyceridemia. When triglycerides exceed 500 mg/dL (pancreatitis risk), niacin is one of several agents (along with fibrates, omega-3) that can rapidly bring triglycerides down.
- Atherogenic dyslipidemia in metabolic syndrome WITHOUT existing statin therapy. Patients with low HDL, high triglycerides, small-dense LDL who are not yet on statins (often because of preference or partial intolerance) can benefit from niacin's spectrum of lipid effects.
- NOT recommended: Adding niacin on top of optimized high-intensity statin + ezetimibe therapy in patients already at LDL goal. AIM-HIGH and HPS2-THRIVE settled this question.
For the broader cardiovascular workup, see Cardiovascular Disease and Lipid Panel.
The Lipoprotein(a) Question
Lipoprotein(a) is an LDL-like particle with an additional apolipoprotein(a) covalently bound. Lp(a) levels are 80-90% genetically determined — you inherit your Lp(a) at birth and it changes little over a lifetime. Elevated Lp(a) (above 50 mg/dL or 125 nmol/L) is an independent risk factor for atherosclerotic cardiovascular disease and aortic stenosis, with the relative risk roughly equivalent to elevated LDL.
The problem: until recently, almost no drug meaningfully lowered Lp(a):
- Statins: no effect or slight increase
- Ezetimibe: no effect
- Niacin: 20-30% reduction — the strongest of the available oral drugs
- PCSK9 inhibitors: 20-30% reduction, similar to niacin but ~1000x more expensive
- Pelacarsen (antisense oligonucleotide): 80% reduction in phase 2 trials; phase 3 outcomes trial (HORIZON) ongoing
- Lipoprotein apheresis: dramatic reduction but requires weekly outpatient treatment, expensive, reserved for severe cases
For a patient with Lp(a) of 100 mg/dL and otherwise well-controlled lipids, niacin remains a defensible option pending the pelacarsen outcomes data. The Lp(a)-lowering effect is one of niacin's most enduring claims to clinical relevance.
Mechanism of Lipid Modification
Niacin's lipid-modifying effects derive from multiple convergent mechanisms:
- Inhibition of adipose triglyceride lipase (ATGL) via GPR109A. GPR109A (also called HCA2 or HCAR2) is the niacin receptor expressed on adipocytes. Activation suppresses lipolysis, reducing free fatty acid (FFA) flux to the liver. Lower hepatic FFA delivery reduces VLDL assembly and secretion, lowering triglycerides and VLDL-derived LDL.
- Inhibition of DGAT2 (diacylglycerol acyltransferase 2) in hepatocytes, reducing triglyceride synthesis and VLDL assembly.
- Inhibition of hepatic ApoB synthesis and increased ApoB intracellular degradation, lowering all ApoB-containing particles (VLDL, IDL, LDL, Lp(a)).
- Increased ApoA-I production in the liver, raising HDL particle number.
- Reduced HDL catabolism through decreased ATP synthase beta-chain receptor activity, extending HDL plasma residence time.
- Reduced Lp(a) production through downregulation of apolipoprotein(a) gene expression in hepatocytes.
The flush is a side effect of GPR109A activation on Langerhans cells in the skin, where it triggers prostaglandin D2 release and consequent vasodilation. This is separate from the adipose GPR109A effect that drives lipid modification — you cannot block the flush without partially blocking the lipid benefit (which is why laropiprant failed in HPS2-THRIVE).
1-3 g/day Immediate-Release Titration Protocol
For appropriately selected patients (see Modern Positioning above), the practical protocol is:
Pre-treatment workup
- Baseline lipid panel including ApoB and Lp(a)
- Baseline ALT, AST, bilirubin
- Baseline fasting glucose and HbA1c
- Baseline uric acid
- Patient education on flush expectation and management
Titration schedule
- Weeks 1-2: 100 mg IR nicotinic acid with the largest meal of the day. Optional aspirin 325 mg 30 minutes before the dose during the first 1-2 weeks to attenuate flushing.
- Weeks 3-4: Increase to 250 mg with the largest meal (or 100 mg twice daily). Reassess flush tolerance.
- Weeks 5-6: 500 mg once daily with the largest meal, or 250 mg twice daily.
- Weeks 7-8: 500 mg twice daily (1 g/day total).
- Weeks 9-12: Titrate further toward 1500-2000 mg/day in 2-3 divided doses with meals if needed to reach lipid targets.
- Maintenance: 1000-2000 mg/day in divided doses for most patients; up to 3000 mg/day in selected cases with rigorous monitoring.
Monitoring schedule
- Baseline, 6 weeks, 12 weeks, then every 6 months: ALT, AST, bilirubin, fasting glucose, HbA1c, uric acid
- Baseline and every 3-6 months: lipid panel (full panel including ApoB, Lp(a))
- Discontinue if: ALT exceeds 3x upper limit of normal, fasting glucose rises > 30 mg/dL, gout flare occurs, persistent intolerable flushing, or new significant adverse effect
Critical formulation choice
Use immediate-release (IR) nicotinic acid only. Brand-name preparations have largely disappeared from the US market; quality generic IR niacin is widely available from reputable supplement manufacturers. Sustained-release (SR) and extended-release (ER) formulations — despite causing less flush — carry substantially higher hepatotoxicity risk and should NOT be used at gram-range doses. The McKenney 1994 JAMA paper directly compared and found dramatic SR-niacin hepatotoxicity at lipid-modifying doses.
Cautions
- HEPATOTOXICITY — sustained-release nicotinic acid at >1 g/day is the single most important contraindication. SR/ER niacin formulations have caused elevated transaminases, cholestatic jaundice, and fulminant hepatic failure at lipid-modifying doses. Use immediate-release only. Monitor liver enzymes per the protocol above. Discontinue at any ALT > 3x ULN.
- Hyperglycemia. Niacin raises fasting glucose by ~5-10 mg/dL and HbA1c by ~0.3-0.4 in many patients via increased lipolysis and free fatty acid release driving hepatic gluconeogenesis. In diabetic patients monitor glucose closely; do not assume the lipid benefit outweighs the glycemic cost. In well-controlled diabetes, the trade-off may favor proceeding; in poorly controlled diabetes, choose a different lipid-modifying strategy.
- Gout and hyperuricemia. Niacin competes with uric acid for renal tubular excretion, raising serum uric acid by 0.5-1.5 mg/dL and potentially triggering gout flares. Avoid in active gout or symptomatic hyperuricemia; in patients with prior gout but currently asymptomatic, co-administer allopurinol or febuxostat.
- Peptic ulcer activation. Niacin increases gastric acid secretion. Avoid in active peptic ulcer disease. Patients with prior PUD currently on PPI or H2 blocker can usually proceed with caution.
- Flush. Universal at gram-range IR doses initially. Manage with slow titration, aspirin pre-treatment, taking with food, and patient education that the flush diminishes substantially over 1-2 weeks of consistent dosing.
- Pregnancy. Pharmacological niacin doses are not established as safe in pregnancy. Avoid unless specifically directed by a maternal-fetal medicine specialist.
- Drug interactions: warfarin (niacin may potentiate — monitor INR more frequently after initiation); statin myopathy risk modestly increased; bile-acid sequestrants reduce niacin absorption (separate dosing by 4 hours).
- Lp(a) target ambiguity. While niacin lowers Lp(a) by 20-30%, no randomized trial has yet shown that Lp(a) lowering by any drug translates into hard cardiovascular outcomes. The pelacarsen HORIZON trial is the key upcoming evidence. Niacin for Lp(a) is currently a mechanistic bet, not a proven outcome.
Key Research Papers
- Coronary Drug Project Research Group (1975). Clofibrate and niacin in coronary heart disease. JAMA. — PubMed
- Canner PL et al. (1986). Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. JACC. — PubMed
- Carlson LA, Rosenhamer G (1988). Reduction of mortality in the Stockholm Ischaemic Heart Disease Secondary Prevention Study by combined treatment with clofibrate and nicotinic acid. Acta Med Scand. — PubMed
- AIM-HIGH Investigators / Boden WE et al. (2011). Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. NEJM. — PubMed
- HPS2-THRIVE Collaborative Group (2014). Effects of extended-release niacin with laropiprant in high-risk patients. NEJM. — PubMed
- Carlson LA (2005). Nicotinic acid: the broad-spectrum lipid drug — a 50th anniversary review. J Intern Med. — PubMed
- Bruckert E et al. (2010). Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis. — PubMed
- McKenney JM et al. (1994). A comparison of the efficacy and toxic effects of sustained- vs immediate-release niacin in hypercholesterolemic patients. JAMA. — PubMed
- Niacin and Lp(a) reduction — PubMed
- Niacin and ApoB lowering — PubMed
- GPR109A receptor and niacin lipid mechanism — PubMed
- Niacin in statin-intolerant patients — PubMed
PubMed Topic Searches
- PubMed: niacin cardiovascular mortality
- PubMed: niacin HDL coronary
- PubMed: nicotinic acid atherosclerosis
- PubMed: niacin hepatotoxicity SR
- PubMed: niacin gout glucose
Connections
- Vitamin B3 Overview
- B3 Benefits Hub
- B3 as NAD+ Precursor
- B3 for Pellagra & Skin
- B3 for Schizophrenia & Mental Health
- Niacin & Cholesterol (Original Article)
- Cardiovascular Disease
- Cholesterol Management
- Atherosclerosis
- Coronary Artery Disease
- Lipid Panel
- Niacin in Gerson Therapy
- Alpha Lipoic Acid
- Uric Acid
- All Vitamins