Autophagy and mTOR — The Molecular Biology of Fasting
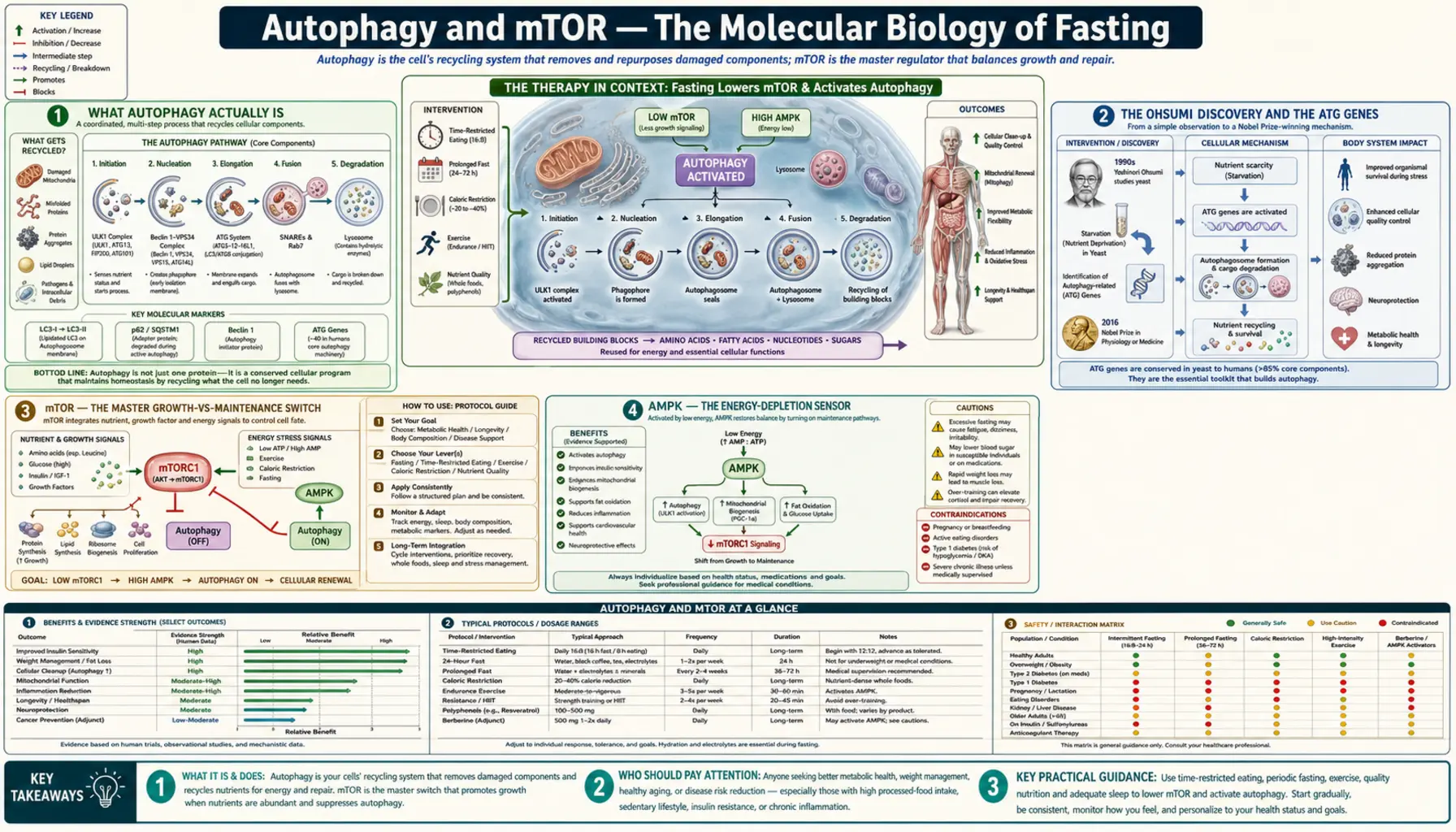
In 2016, the Nobel Prize in Physiology or Medicine was awarded to Yoshinori Ohsumi for his discoveries of the molecular machinery of autophagy — the cellular self-cleaning process by which cells degrade and recycle their own damaged components. Ohsumi's yeast-genetic screens in the 1990s identified the ATG (autophagy-related) genes that encode the membrane-trafficking machinery for the formation of autophagosomes — the double-membrane vesicles that engulf damaged organelles, misfolded proteins, and even invading intracellular pathogens, deliver them to the lysosome for degradation, and recycle the molecular components as amino acids, fatty acids, and nucleotides. Autophagy is the cellular equivalent of a recycling program. It is the most powerful endogenous mechanism humans possess for managing the accumulated damage of aging, and fasting is the single most powerful physiological inducer of it. The control system — mTOR signaling — is what determines moment-by-moment whether a cell is in growth-and-synthesis mode or maintenance-and-recycling mode.
Interactive Visualization Fat Burning & Ketosis — what actually happens Follow a fatty acid out of a fat cell to the mitochondrial gate and find it padlocked by insulin. Drop the carbs, open CPT1, and watch the liver start making ketones. Launch →
Table of Contents
- What Autophagy Actually Is
- The Ohsumi Discovery and the ATG Genes
- mTOR — The Master Growth-vs-Maintenance Switch
- AMPK — The Energy-Depletion Sensor
- Leucine and Amino-Acid mTOR Activation
- Insulin, IGF-1, and PI3K/Akt Branch
- Selective Autophagy (Mitophagy, Pexophagy, Xenophagy)
- Disease Implications — Cancer, Neurodegeneration, Aging
- How to Maximize Autophagy in Practice
- Key Research Papers
- Connections
- Featured Videos
What Autophagy Actually Is
Cells contain organelles that wear out, proteins that misfold, lipid droplets that accumulate, and the occasional intracellular pathogen. Without a mechanism to dispose of and recycle this damaged material, cells would slowly fill with debris, lose function, and eventually die — which is essentially what happens in aging tissues, and what happens dramatically in neurodegenerative diseases where damaged proteins (amyloid-beta in Alzheimer's, alpha-synuclein in Parkinson's, mutant huntingtin in Huntington's) accumulate in neurons.
Autophagy — from the Greek auto ("self") + phagein ("to eat") — is the cell's solution. There are three distinct mechanisms:
- Macroautophagy — the most studied form. A flat membrane sheet (the phagophore) extends around a piece of cytoplasm containing damaged organelles or misfolded proteins, fuses with itself to form a double-membrane vesicle called an autophagosome, and then fuses with a lysosome where acid hydrolases degrade the cargo. The amino acids, fatty acids, and nucleotides released by degradation are recycled back to the cell for the synthesis of fresh components. This is what most people mean by "autophagy" in the popular fasting literature.
- Microautophagy — the lysosomal membrane directly invaginates to engulf small cytoplasmic contents. Less well characterized.
- Chaperone-mediated autophagy (CMA) — specific proteins bearing a KFERQ motif are recognized by the cytosolic chaperone HSC70 and delivered to the lysosome via the LAMP-2A receptor. This is a targeted disposal mechanism for specific proteins, not a bulk recycling system.
All three are upregulated during fasting and downregulated during the fed state. The macroautophagy response to fasting is the most dramatic — autophagic flux can increase 4-10-fold in liver, heart, and muscle tissue within 24-48 hours of fasting in mouse models. Human data are more limited because measuring autophagy in human tissue requires biopsy, but indirect surrogate markers (serum LC3-II, autophagy-related gene expression in PBMCs) show parallel changes.
The Ohsumi Discovery and the ATG Genes
Yoshinori Ohsumi's laboratory at the University of Tokyo began studying autophagy in the late 1980s, when the basic phenomenon of vacuolar protein degradation was known but the molecular machinery was completely opaque. Ohsumi made the brilliant methodological decision to study autophagy in baker's yeast (Saccharomyces cerevisiae) — an organism that could be subjected to systematic genetic screens.
His 1992 Journal of Cell Biology paper described starved yeast cells accumulating autophagic bodies in their vacuoles, visible under light microscopy when vacuolar protease activity was blocked. With this assay, his lab conducted a mutagenesis screen and identified 15 yeast genes required for autophagy — the original ATG genes (atg1 through atg15). Subsequent work expanded the list to over 40 ATG genes and identified the mammalian homologs.
The molecular cascade Ohsumi and others worked out:
- Initiation — The ULK1 kinase complex (ATG1 in yeast) is the most upstream regulator. It is activated by AMPK phosphorylation when energy is low, and inhibited by mTOR phosphorylation when growth signaling is active. This reciprocal regulation is the molecular switch.
- Nucleation — The class III PI3K complex (containing Beclin-1 / ATG6, VPS34, and ATG14L) produces phosphatidylinositol 3-phosphate (PI3P) at the phagophore assembly site.
- Elongation — Two ubiquitin-like conjugation systems (ATG12-ATG5-ATG16L1 and LC3-PE) extend the phagophore membrane. The LC3-II form (PE-conjugated LC3) is the canonical molecular marker of autophagosome formation used in research.
- Closure and fusion — The autophagosome closes into a double-membrane vesicle and traffics to the lysosome, where SNARE-mediated fusion produces the autolysosome and cargo degradation.
Every step is conserved from yeast through mammals — a striking testament to the evolutionary antiquity of the autophagy machinery and its centrality to eukaryotic cell biology.
mTOR — The Master Growth-vs-Maintenance Switch
mTOR (mechanistic target of rapamycin, formerly "mammalian target of rapamycin") is a serine/threonine kinase that integrates signals about nutrient sufficiency, energy state, growth factors, and oxygen availability, and determines whether the cell will pursue growth-and-proliferation or maintenance-and-recycling.
mTOR exists in two functionally distinct complexes:
- mTORC1 — contains mTOR, Raptor, mLST8, PRAS40, and DEPTOR. mTORC1 is the nutrient-sensing complex that responds to amino acids (especially leucine), glucose, growth factors (via PI3K/Akt), and energy state (via AMPK). When active, mTORC1 phosphorylates S6K1 to drive ribosome biogenesis and 4E-BP1 to release the eIF4E translation initiation factor — together driving protein synthesis. mTORC1 simultaneously phosphorylates and inhibits ULK1, blocking autophagy initiation. Rapamycin is a specific mTORC1 inhibitor (in short exposure; chronic rapamycin also inhibits mTORC2).
- mTORC2 — contains mTOR, Rictor, mLST8, mSin1, and Protor. mTORC2 responds to growth factors and is the activator of Akt (phosphorylates Akt-Ser473). It plays roles in cell survival, cytoskeletal organization, and metabolism. mTORC2 is less directly involved in autophagy regulation and is not the primary fasting-responsive complex.
The fed state — when blood glucose, insulin, IGF-1, and circulating amino acids (especially leucine) are all elevated — pushes mTORC1 to maximal activity. Protein synthesis runs. Autophagy is suppressed. The cell builds. The fasted state — when all four signals decline — releases mTORC1 inhibition of ULK1. Autophagy initiates. The cell cleans house and recycles its components.
This reciprocal architecture is why fasting is the most powerful physiological autophagy inducer. There is no supplement or drug short of rapamycin itself that produces the equivalent simultaneous downregulation of all four mTORC1 inputs.
AMPK — The Energy-Depletion Sensor
If mTOR is the nutrient-sensing master, AMP-activated protein kinase (AMPK) is the energy-state sensor that operates as its functional opposite. AMPK is allosterically activated by AMP and ADP — the products of ATP hydrolysis. When cellular ATP is being used faster than it is being produced, the AMP:ATP ratio rises, and AMPK switches on.
Activated AMPK has three major actions relevant to fasting:
- Direct ULK1 activation — AMPK phosphorylates ULK1 at Ser317 and Ser777, directly activating it and overriding the mTORC1 inhibition. This is the direct AMPK-driven path to autophagy.
- Indirect mTORC1 inhibition — AMPK phosphorylates and activates TSC2, which converts Rheb-GTP to Rheb-GDP and thereby inhibits mTORC1. AMPK also directly phosphorylates Raptor (within mTORC1), causing 14-3-3 binding and further mTORC1 inhibition.
- Metabolic reprogramming — AMPK phosphorylates and inhibits acetyl-CoA carboxylase (blocking fatty-acid synthesis), activates carnitine palmitoyltransferase 1 (driving fatty acid oxidation), activates GLUT4 translocation (increasing glucose uptake in muscle), and increases mitochondrial biogenesis through PGC-1alpha.
The drug metformin (the front-line type-2-diabetes therapy) activates AMPK as its proposed primary mechanism, which is why metformin produces some of the same metabolic signatures as fasting — improved insulin sensitivity, increased fat oxidation, modest autophagy induction. The longevity-research interest in metformin (the TAME trial) is essentially the hypothesis that pharmacological AMPK activation could partially replicate the fasting longevity signal.
Exercise — particularly intense exercise — also activates AMPK by transiently elevating the AMP:ATP ratio. This is part of why the combination of intermittent fasting and exercise produces synergistic effects on metabolic health — both interventions independently activate AMPK, and the effects compound.
Leucine and Amino-Acid mTOR Activation
Among the inputs to mTORC1, amino acids — and particularly the branched-chain amino acid leucine — have an outsized regulatory effect. Leucine binds to the cytosolic sensor sestrin2, releasing sestrin's inhibition of GATOR2, which permits Rag GTPase-mediated recruitment of mTORC1 to the lysosomal surface where it can be activated by Rheb.
Practical implication: leucine breaks a fast in terms of mTOR signaling, even in trivial amounts. A small protein snack of 5-10 grams of complete protein (about 1 g of leucine) is sufficient to reactivate mTORC1 and shut down autophagy. This is why "just a little protein" during a fasting window is not metabolically trivial — it specifically eliminates the autophagy benefit even if the calorie content is negligible.
Conversely, ketogenic diets and very-low-protein diets (the rationale behind some fasting-mimicking diet protocols) keep leucine low even while providing some calories, allowing mTORC1 suppression to persist. This is why the FMD is designed to be 9-11% protein — low enough to keep mTORC1 mostly off while providing enough food intake to improve safety and tolerability.
The corollary for muscle health: leucine is also the most powerful trigger of muscle protein synthesis. The leucine threshold of approximately 2.5-3 g per meal is required to maximally stimulate MPS in healthy young adults; older adults need approximately 3-4 g per meal due to anabolic resistance. For practitioners trying to combine fasting with muscle maintenance, the strategy is to time the fed-window meals to deliver supraphysiologic leucine pulses (which efficiently support MPS within the 8-hour window) while keeping the fasting window strictly leucine-free.
Insulin, IGF-1, and PI3K/Akt Branch
The second major fed-state mTOR input is the insulin / IGF-1 / PI3K / Akt growth-factor signaling axis. Insulin and IGF-1 bind their respective receptor tyrosine kinases on the cell surface. The activated receptors recruit insulin receptor substrate (IRS1/2), which activates phosphoinositide 3-kinase (PI3K). PI3K phosphorylates membrane PIP2 to PIP3, which recruits Akt (also called PKB). PDK1 then phosphorylates Akt at Thr308 and mTORC2 phosphorylates Akt at Ser473, activating Akt.
Activated Akt phosphorylates and inactivates TSC2 (the inhibitor of mTORC1 noted above). With TSC2 inactivated, Rheb-GTP accumulates and activates mTORC1. The PI3K/Akt/mTORC1 cascade is the dominant signaling pathway that links systemic anabolism (insulin from a meal, IGF-1 from growth hormone action) to cell-autonomous protein synthesis.
The longevity implication: lifelong reduction in IGF-1 signaling is associated with extended lifespan across species from C. elegans (the daf-2 / age-1 insulin/IGF-1 pathway mutants discovered by Cynthia Kenyon) through mice (long-lived Ames dwarf and Snell dwarf mutants have low IGF-1) to humans (Laron syndrome patients with congenital growth-hormone receptor deficiency have strikingly low cancer incidence, per Guevara-Aguirre and Longo's Ecuadorian cohort).
Fasting acutely lowers insulin and IGF-1 and chronically reduces fasting insulin via improved insulin sensitivity, intersecting with the same longevity-associated signaling reductions. The fasting / caloric-restriction longevity literature and the IGF-1 longevity literature are essentially studying the same fundamental phenomenon from different methodological angles.
Selective Autophagy (Mitophagy, Pexophagy, Xenophagy)
Bulk autophagy degrades cytoplasmic material indiscriminately. But the autophagy machinery can also be selectively targeted to specific cargo, producing several specialized clearance systems:
- Mitophagy — the selective autophagy of damaged or depolarized mitochondria. The PINK1 / Parkin pathway is the most studied: PINK1 is normally imported into healthy mitochondria and degraded; in depolarized mitochondria, PINK1 accumulates on the outer membrane and recruits Parkin, which ubiquitinates outer-membrane proteins and tags the mitochondrion for autophagic clearance. Mitophagy is critically important in neurons (high mitochondrial demand, post-mitotic cells) and is impaired in Parkinson's disease — PINK1 and PARK2 mutations cause familial Parkinson's.
- Pexophagy — selective clearance of peroxisomes, important during developmental transitions.
- ER-phagy — clearance of damaged or excess endoplasmic reticulum, important after ER stress.
- Xenophagy — the cell's targeted autophagic destruction of intracellular pathogens. Group A streptococcus, Salmonella, Mycobacterium tuberculosis, and herpes simplex virus all face the xenophagy response when they enter the cytoplasm. Several pathogens have evolved counter-mechanisms to block autophagy — one of the host-pathogen molecular arms races.
- Aggrephagy — clearance of misfolded protein aggregates. Particularly relevant to neurodegeneration: aggrephagy of amyloid-beta, alpha-synuclein, mutant huntingtin, and TDP-43 inclusions.
- Lipophagy — autophagic degradation of lipid droplets in hepatocytes and adipocytes. Important in fatty liver disease.
Fasting upregulates all of these selective autophagy pathways, not just the bulk macroautophagy. Mitophagy in particular is dramatically increased — one of the proposed mechanisms by which fasting improves mitochondrial quality and function in aged tissue.
Disease Implications — Cancer, Neurodegeneration, Aging
- Cancer — autophagy plays a Janus-faced role in cancer. In early/pre-cancerous cells, robust autophagy clears damaged organelles and reduces oxidative stress, suppressing the genomic instability that drives transformation. In established tumors with constitutively activated mTOR (PI3K/Akt/mTOR is one of the most commonly hyperactivated pathways in human cancer), autophagy can protect tumor cells from stress and chemotherapy. The therapeutic implications differ by context: autophagy induction may be cancer-preventive, while autophagy inhibition (chloroquine, hydroxychloroquine in clinical trials) may sensitize established tumors to chemotherapy. The fasting-as-chemotherapy-adjunct work discussed on the extended-fasts page exploits the cancer-cell inability to downshift mTOR in response to nutrient deprivation.
- Neurodegeneration — Alzheimer's disease, Parkinson's disease, Huntington's disease, and ALS are all characterized by accumulation of misfolded proteins that should normally be cleared by autophagy. Aging itself reduces baseline autophagic flux in many tissues including the brain. The hypothesis that fasting-induced autophagy could clear damaged proteins and slow neurodegenerative progression is biologically plausible and is being tested in multiple clinical trials. Direct evidence in humans is still preliminary, but the mouse-model evidence is strong.
- Aging — autophagic flux declines with age in most tissues. The accumulation of damaged organelles, particularly mitochondria, is one of the hallmarks of cellular senescence. Interventions that restore autophagy (caloric restriction, fasting, rapamycin, the polyamine spermidine) extend lifespan in multiple model organisms.
- Inflammation and autoimmunity — autophagy controls inflammatory signaling through several mechanisms, including the NLRP3 inflammasome (suppressed by mitophagy clearing damaged mitochondria that would otherwise activate NLRP3). Defects in autophagy genes (ATG16L1) are associated with inflammatory bowel disease, particularly Crohn's disease.
- Cardiac protection — autophagy in cardiomyocytes is essential for cellular quality control. Fasting and exercise both upregulate cardiac autophagy. Several studies link improved cardiac autophagy to better recovery from ischemic injury.
How to Maximize Autophagy in Practice
- Extended fasting (24-72 hours) — the most powerful physiological autophagy inducer. See the extended-fasts page for protocols.
- Daily time-restricted eating with the longest tolerable fasting window — 16:8 produces modest autophagy; tighter windows produce more.
- Ketogenic diet — chronic carbohydrate restriction with adequate fat keeps insulin and mTOR low even in the fed state, producing modest baseline autophagy upregulation.
- Strict avoidance of leucine during the fasting window — no protein, no broth with meat protein, no BCAA supplements during the fast. Even a few grams of leucine reactivates mTORC1 and shuts down autophagy.
- Exercise during the fast — particularly fasted aerobic and resistance exercise. AMPK activation from exercise compounds with the fasting-induced AMPK activation.
- Coffee and tea — coffee in particular is a modest autophagy inducer through caffeine and chlorogenic acid effects on AMPK and mTOR. Compatible with fasting.
- Spermidine-rich foods during the fed window — wheat germ, aged cheese, and fermented soy products are concentrated sources of spermidine, a polyamine that induces autophagy and has been shown to extend lifespan in multiple species.
- Avoid frequent snacking — even a small calorie-bearing snack interrupts the fasting state and reactivates mTOR. Three discrete meals in an 8-10 hour window is metabolically better than 5-6 small snacks across the day, even at identical total calories.
Key Research Papers
- Ohsumi Y (2014). Historical landmarks of autophagy research. Cell Research. — PubMed
- Mizushima N, Komatsu M (2011). Autophagy: renovation of cells and tissues. Cell. — PubMed
- Saxton RA, Sabatini DM (2017). mTOR signaling in growth, metabolism, and disease. Cell. — PubMed
- Hardie DG, Ross FA, Hawley SA (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature Reviews Molecular Cell Biology. — PubMed
- Kim J et al. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology. — PubMed
- Sancak Y et al. (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. — PubMed
- Wolfson RL et al. (2016). Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. — PubMed
- Levine B, Kroemer G (2019). Biological functions of autophagy genes: a disease perspective. Cell. — PubMed
- Madeo F et al. (2018). Spermidine in health and disease. Science. — PubMed
- Harrison DE et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. — PubMed
- Pickrell AM, Youle RJ (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. — PubMed
- Bjedov I et al. (2010). Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metabolism. — PubMed
PubMed Topic Searches
- PubMed: Autophagy and fasting
- PubMed: mTORC1 leucine sensing
- PubMed: AMPK and autophagy
- PubMed: Mitophagy PINK1/Parkin
- PubMed: Rapamycin and longevity
Connections
- Fat Burning & Ketosis — interactive animation
- Fasting Benefits Hub
- Fasting Overview
- Time-Restricted Eating 16:8
- Extended Fasts (24-72h)
- Refeeding Strategy
- L-Leucine (mTOR Trigger)
- Alzheimer's Disease
- Parkinson's Disease
- Oncology
- Fatty Liver Disease
- Type 2 Diabetes
- Insulin Resistance
- All Remedies
- Eggs (Leucine Source)