Chromium for Insulin Sensitivity
Insulin sensitivity — the capacity of peripheral tissues to respond to a given concentration of circulating insulin — is the single most important hinge in chromium biology. Every other benefit ascribed to chromium (glycemic control, triglyceride reduction, modest weight effects, reduction in carbohydrate cravings, possible cardiovascular protection) flows downstream from one molecular event: holo-chromodulin binding to the activated insulin receptor and amplifying its tyrosine kinase activity by up to eight-fold in vitro. This deep-dive walks through the bioavailability differences between chromium picolinate, polynicotinate, chloride, and histidinate; the decades of mechanistic and clinical work done at the USDA Beltsville Human Nutrition Research Center under Richard Anderson; the discovery and structural characterization of the low-molecular-weight chromium-binding substance (LMWCr / chromodulin) by John Vincent and colleagues; the AS160-GLUT4 translocation pathway in skeletal muscle; and the practical clinical translation for non-diabetic insulin-resistant populations including PCOS, prediabetes, and metabolic syndrome.
Table of Contents
- What "Insulin Sensitivity" Actually Means
- Forms: Picolinate vs Polynicotinate vs Chloride vs Histidinate
- Richard Anderson's USDA Beltsville Research Program
- Chromodulin (LMWCr) — Discovery, Structure, Function
- Insulin Receptor Tyrosine Kinase Phosphorylation
- IRS-1/IRS-2, PI3K, Akt, and AS160
- GLUT4 Translocation in Skeletal Muscle and Adipose
- Transferrin-Mediated Chromium Delivery
- Non-Diabetic Insulin-Resistant Populations (PCOS, Prediabetes)
- Cautions and the Skeptical View
- Key Research Papers
- Connections
- Featured Videos
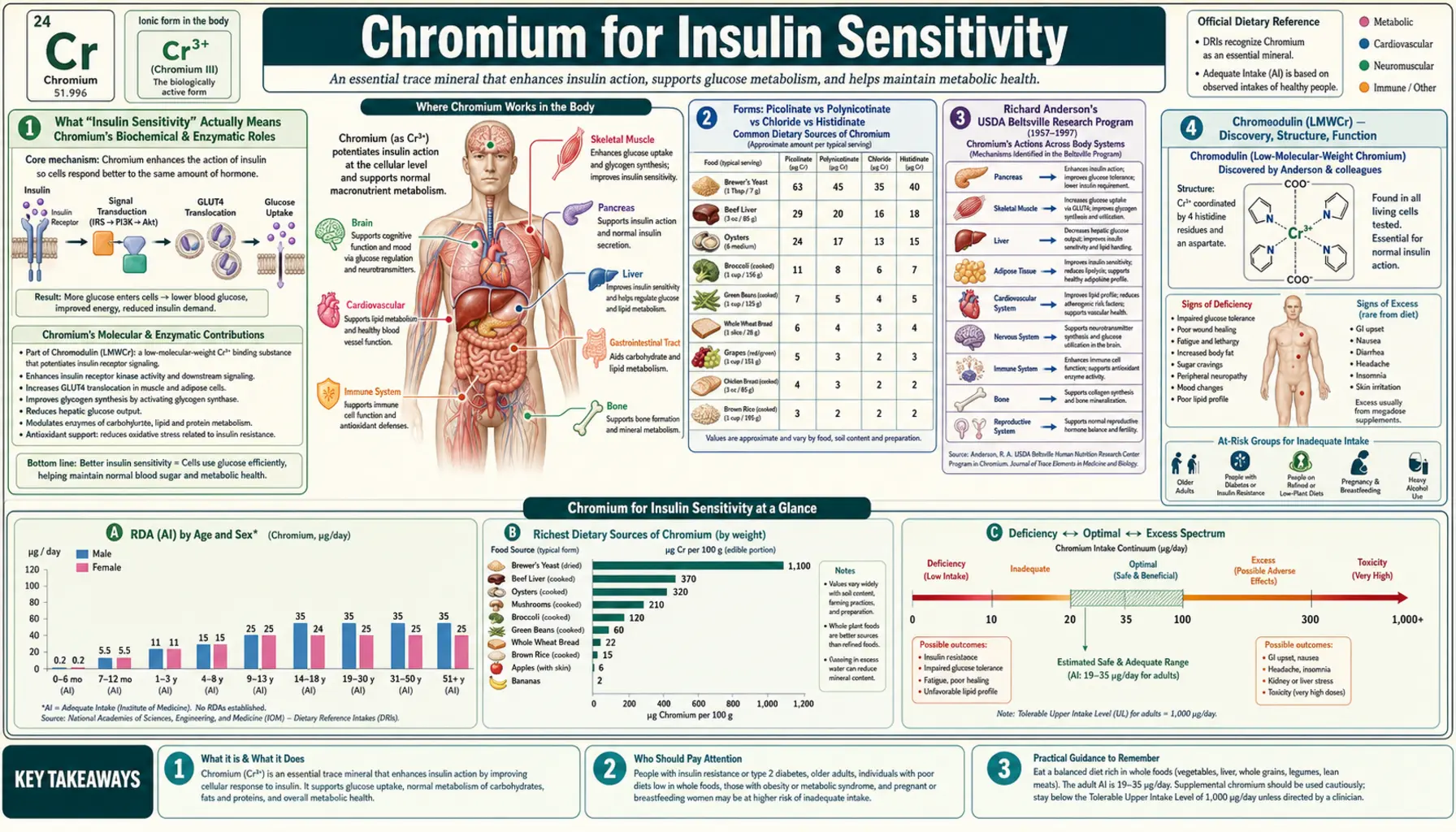
What "Insulin Sensitivity" Actually Means
The clinical literature uses "insulin sensitivity" and "insulin resistance" as if their meanings were obvious, but the underlying physiology is worth pinning down precisely before discussing how chromium influences it. Insulin sensitivity is the inverse of insulin resistance: a measure of how much intracellular signal a given concentration of insulin produces at its target tissues, most importantly skeletal muscle, liver, and adipose tissue.
The gold-standard quantitative measure is the hyperinsulinemic-euglycemic clamp, developed by Ralph DeFronzo at Yale in 1979. In a clamp study, insulin is infused at a fixed rate while glucose is infused at whatever rate is needed to keep blood glucose constant. The glucose infusion rate (M-value, expressed as mg/kg/min) is the direct measure of whole-body insulin-stimulated glucose disposal — higher numbers mean greater insulin sensitivity. Most clinical work uses cheaper proxies: HOMA-IR (fasting glucose times fasting insulin, divided by 22.5), the Matsuda index from an oral glucose tolerance test, or simply fasting insulin alone.
Insulin resistance precedes type 2 diabetes by years or decades. As muscle, liver, and adipose tissue become progressively less responsive to insulin, the pancreatic beta cells compensate by secreting more insulin to maintain normoglycemia — this is the compensatory hyperinsulinemia phase, where fasting insulin rises but fasting glucose remains in the normal range. Eventually, the beta cells exhaust their compensatory capacity (a combination of beta-cell apoptosis, dedifferentiation, and lipotoxic damage from chronically elevated free fatty acids), insulin secretion drops relative to need, and frank type 2 diabetes emerges. Insulin sensitization is therefore not just diabetes treatment — it is upstream prevention.
This is the conceptual lens through which to read the chromium evidence. The Balk 2007 Diabetes Care meta-analysis finding that chromium helps people with diagnosed diabetes but not people without diabetes is not actually a contradiction: it reflects the fact that insulin sensitization produces measurable downstream benefits (HbA1c reduction, fasting glucose reduction) most easily in tissues whose insulin signaling is already failing. In a healthy adult with normal insulin sensitivity, amplifying an already-adequate signal produces little visible change.
Forms: Picolinate vs Polynicotinate vs Chloride vs Histidinate
The choice of chromium form matters enormously for bioavailability and clinical outcome. The four commonly encountered forms differ by an order of magnitude or more in intestinal absorption efficiency.
- Chromium picolinate — chromium chelated to three picolinate (2-pyridinecarboxylate) molecules. This is the most-studied form in human clinical trials, used in roughly 80% of published chromium intervention studies, and the form Anderson chose for the landmark USDA Beltsville Chinese type 2 diabetes trial. Picolinic acid is a tryptophan metabolite that the small intestine actively transports, and it carries the chelated chromium into enterocytes by hitchhiking on its own absorption mechanism. Bioavailability is typically estimated at 1.2–2.8% — low in absolute terms but several-fold higher than chromium chloride. The picolinate moiety also enhances cellular uptake from the bloodstream into target tissues. Typical research dose: 200–1,000 mcg/day.
- Chromium polynicotinate (niacin-bound chromium) — chromium complexed with three niacin (nicotinic acid) molecules. This formulation was designed to mimic the structure of "glucose tolerance factor" (GTF), the naturally occurring chromium-niacin complex first isolated from brewer's yeast by Walter Mertz and colleagues at USDA in the late 1950s. Bioavailability is comparable to picolinate, and head-to-head trials show similar effects on glucose and lipid markers. Marketed under brand names such as ChromeMate. Some marketing claims that polynicotinate is "safer" than picolinate — the safety differential, if any, is small and clinically irrelevant at standard doses.
- Chromium chloride (CrCl3) — the inorganic salt. Bioavailability is roughly 0.4%, three to five times lower than chelated organic forms. CrCl3 was the form used in early TPN repletion case reports (where intravenous administration bypasses the absorption problem entirely) and in older clinical trials. Modern supplements rarely use it. The fact that it works at all in the TPN setting confirms that chromium itself, not the chelator, is the active species — the picolinate and nicotinate carriers exist to solve the absorption problem, not to add independent biological activity.
- Chromium histidinate — chromium chelated to the amino acid histidine. A newer form with promising in vitro and animal data, plus a small number of human trials suggesting comparable or slightly superior bioavailability to picolinate. Cost is higher and the human evidence base remains limited.
- Brewer's yeast (food-source GTF) — whole brewer's yeast contains naturally complexed chromium in the GTF form, plus B vitamins and other cofactors. Concentration is variable (some yeasts are essentially chromium-free; others contain meaningful amounts). Food-source chromium has the advantage of physiologic dosing and the disadvantage of inconsistent content.
For a clinical recommendation: chromium picolinate at 200–500 mcg/day is the form with the strongest evidence base, the most predictable absorption, and acceptable cost. Chromium polynicotinate is an equivalent alternative. Chromium chloride is obsolete for oral supplementation. Chromium histidinate is reasonable for patients who specifically request a picolinate alternative.
Richard Anderson's USDA Beltsville Research Program
Almost every major piece of chromium clinical evidence traces back, directly or indirectly, to the USDA Beltsville Human Nutrition Research Center in Maryland and the four-decade research program led by Richard A. Anderson, PhD. Anderson and his colleagues at Beltsville published more than 250 peer-reviewed papers on chromium biology between the 1970s and his retirement in the 2010s, covering everything from the molecular biology of chromodulin to the clinical pharmacology of chromium picolinate in poorly controlled type 2 diabetes.
The single most-cited Anderson paper is the 1997 Diabetes publication of the Beijing trial: 180 Chinese adults with type 2 diabetes randomized in a four-month, double-blind, placebo-controlled trial of three doses of chromium picolinate (0 / 200 / 1,000 mcg/day, two pills BID). The 1,000 mcg/day group showed:
- HbA1c reduction of approximately 1.9 percentage points (8.5% → 6.6%)
- Fasting blood glucose reduction of approximately 30 mg/dL
- 2-hour postprandial glucose reduction of approximately 70 mg/dL
- Reduced fasting and 2-hour insulin levels (consistent with improved insulin sensitivity, not increased insulin secretion)
- Reduced total cholesterol
- No adverse effects
The 200 mcg/day group showed smaller but still statistically significant improvements; placebo showed no change. The dose-response relationship was clear, and the magnitude of effect in the high-dose arm rivaled some oral hypoglycemic medications. This trial established 1,000 mcg/day chromium picolinate as the de facto clinical dose for poorly controlled type 2 diabetes, and it remains the headline citation for chromium supplementation more than 25 years later.
Subsequent trials in Western populations have produced more variable results. The general pattern: trials in subjects with poor baseline glycemic control (HbA1c above 8%) replicate the Beijing findings reasonably well; trials in better-controlled diabetics or in prediabetic subjects find smaller and inconsistent effects. The Balk meta-analysis attempted to pool this heterogeneous evidence and reported a mean HbA1c reduction of approximately 0.6 percentage points across all diabetic subjects — smaller than the Beijing trial but clinically meaningful, and statistically robust.
Anderson's other major contributions include the population studies showing that the chromium content of the typical American diet (~33 mcg/day for men, ~25 mcg/day for women) falls below the 50–200 mcg/day Estimated Safe and Adequate Daily Dietary Intake range that was in effect in the 1980s, the demonstration that urinary chromium excretion rises substantially in response to high-glycemic-load meals (suggesting chromium "drain" from chronic carbohydrate excess), and the characterization of chromium losses during exercise (which informs the use of chromium supplementation by athletes).
Chromodulin (LMWCr) — Discovery, Structure, Function
The biological mechanism by which chromium amplifies insulin signaling was a mystery for decades. Walter Mertz had postulated a "glucose tolerance factor" in the late 1950s, a chromium-containing complex found in brewer's yeast that restored normal glucose tolerance in chromium-deficient rats. Subsequent attempts to isolate and characterize GTF were unsuccessful — the active species turned out to be unstable, easily oxidized, and difficult to purify.
The modern understanding emerged from work by John B. Vincent and colleagues at the University of Alabama in the 1990s and 2000s. Vincent isolated and partially characterized a low-molecular-weight chromium-binding substance (initially called LMWCr, later renamed chromodulin) from rabbit liver and bovine colostrum. Key structural features:
- Approximate molecular weight — 1,500 daltons (slightly larger than originally estimated)
- Composition — four amino acid types only: glycine, cysteine, glutamate, and aspartate
- Chromium binding — four trivalent chromium ions per peptide, arranged in a tetranuclear assembly resembling oxo-bridged metal clusters
- Activation requirement — all four chromium ions must be bound for full activity; partially loaded chromodulin (one, two, or three chromium ions) shows progressively reduced insulin-receptor-amplifying activity
The Vincent group's most important functional demonstration: holo-chromodulin (four-chromium loaded) added to isolated rat adipocyte membranes amplified insulin-stimulated insulin receptor tyrosine kinase activity by approximately eight-fold. Apo-chromodulin (no chromium) was inactive. The amplification was specific to the activated receptor — chromodulin did not stimulate the receptor in the absence of insulin, consistent with a role as a signal-amplification cofactor rather than an insulin-mimetic.
The activation cycle, as currently understood:
- In the basal state, apo-chromodulin sits in the cytoplasm of insulin target cells, awaiting chromium delivery
- Insulin binds the receptor; the receptor autophosphorylates; activation signal propagates inward
- Chromium is mobilized from serum transferrin via receptor-mediated endocytosis into the same cells
- Cytoplasmic chromium loads onto apo-chromodulin, converting it to active holo-chromodulin
- Holo-chromodulin binds to the intracellular kinase domain of the activated insulin receptor beta subunit
- Receptor tyrosine kinase activity is amplified up to eight-fold; downstream signaling (IRS → PI3K → Akt → AS160 → GLUT4) is enhanced
- When insulin disengages from the receptor and the signal terminates, chromodulin releases its chromium
- The four chromium ions are excreted in urine; each cycle of insulin signaling effectively consumes a small quantity of chromium
Two clinical inferences follow from this mechanism. First, conditions of chronic hyperinsulinemia (insulin resistance, obesity, sustained high-glycemic-load diet) drive accelerated urinary chromium loss, which is the biochemical basis for the observation that chromium status is often marginal in exactly the populations most likely to benefit from supplementation. Second, the mechanism is genuinely physiologic at low intracellular chromium concentrations — chromodulin's role is to amplify an existing signal, not to produce one from nothing, which is consistent with the clinical observation that chromium supplementation does not cause hypoglycemia in non-diabetic adults.
Insulin Receptor Tyrosine Kinase Phosphorylation
The insulin receptor is a tetramer: two alpha subunits sit on the extracellular face of the cell membrane and bind insulin; two beta subunits span the membrane and project an intracellular kinase domain. When insulin binds the alpha subunits, the receptor undergoes a conformational change that brings the two beta subunits into proximity, allowing each beta subunit to phosphorylate the other on multiple tyrosine residues (Tyr1158, Tyr1162, Tyr1163, and others). This trans-autophosphorylation is the first intracellular event of insulin signaling and the rate-limiting step that determines how strong a signal propagates downstream.
Chromodulin binds to the kinase domain after autophosphorylation has begun. The binding sustains the active conformation, slows the dephosphorylation rate (by reducing access of phosphotyrosine phosphatases such as PTP1B), and prolongs the window during which the receptor can phosphorylate its downstream substrates. The net effect is an approximately three- to eight-fold increase in cumulative tyrosine kinase activity per insulin-binding event, depending on the in vitro assay conditions.
This is mechanistically distinct from how thiazolidinedione drugs (pioglitazone, rosiglitazone) improve insulin sensitivity. Thiazolidinediones act as PPAR-gamma agonists at the transcriptional level, increasing the synthesis of adiponectin and several genes related to fatty acid storage. They take weeks to produce maximal effect because their mechanism requires changes in gene expression. Chromium, in contrast, acts at the post-translational level on receptors that already exist, and its effects on glucose disposal can be measurable within days of starting supplementation in a deficient individual.
The PTP1B angle is interesting because PTP1B is itself a drug target — PTP1B inhibitors are in development as insulin sensitizers. Chromodulin's protective effect on receptor phosphorylation may be loosely analogous to a mild, endogenous PTP1B-inhibitory mechanism. This is one reason combining chromium with other insulin sensitizers (berberine, alpha-lipoic acid, magnesium, cinnamon polyphenols) sometimes produces additive effects in small clinical trials — they act through complementary mechanisms on the same signaling axis.
IRS-1/IRS-2, PI3K, Akt, and AS160
Downstream of the insulin receptor, the canonical insulin signaling cascade flows through:
- Insulin receptor substrate proteins (IRS-1, IRS-2) — large scaffolding proteins phosphorylated by the receptor on multiple tyrosine residues, creating docking sites for downstream SH2-domain-containing effectors
- Phosphoinositide 3-kinase (PI3K) — binds phospho-IRS, becomes activated, and converts membrane PIP2 to PIP3
- PDK1 and Akt (PKB) — recruited to the membrane by PIP3, phosphorylated and activated; Akt is the major effector kinase that propagates the insulin signal to downstream metabolic targets
- AS160 (TBC1D4) — a Rab-GTPase-activating protein that, in its unphosphorylated state, holds GLUT4-containing vesicles in their intracellular storage compartment; once phosphorylated by Akt, AS160 releases its inhibition and allows GLUT4 vesicles to traffic to and fuse with the plasma membrane
- GLUT4 translocation — GLUT4 transporters exposed on the cell surface mediate facilitated glucose entry, completing the cycle from extracellular insulin signal to intracellular glucose disposal
Chromium's amplification of insulin receptor activity propagates through every downstream step of this cascade. In chromium-replete cells, phospho-IRS-1 levels rise, PI3K activity rises, Akt phosphorylation at Ser473 and Thr308 rises, AS160 phosphorylation rises, and GLUT4 surface presentation rises — all proportional to the magnitude of receptor-level amplification. This is why a single mechanism at the most upstream step produces measurable effects on multiple downstream pathways.
One nuance: insulin signaling also flows through a MAPK (mitogen-activated protein kinase) branch that is largely independent of the PI3K/Akt metabolic branch. The MAPK branch drives proliferative, growth-promoting effects of insulin and is the pathway implicated in the proliferative complications of hyperinsulinemia (atherosclerosis, certain cancers). Chromium amplifies the receptor itself, so both branches are theoretically affected, but the literature is much stronger on the metabolic branch. There is no good clinical evidence that chromium supplementation promotes any of the proliferative complications associated with endogenous hyperinsulinemia.
GLUT4 Translocation in Skeletal Muscle and Adipose
The terminal effector of insulin-mediated glucose disposal is GLUT4, a facilitative glucose transporter encoded by the SLC2A4 gene, expressed predominantly in skeletal muscle, cardiac muscle, and adipose tissue. In the basal state, the vast majority of cellular GLUT4 sits inside small intracellular vesicles. Insulin signaling causes these vesicles to migrate to the plasma membrane and fuse with it, presenting GLUT4 transporters at the cell surface where they can transport glucose down its concentration gradient into the cell.
Skeletal muscle is responsible for 70–80% of postprandial insulin-stimulated glucose disposal in a healthy adult. Adipose tissue contributes a smaller but metabolically significant fraction. Improving the efficiency of GLUT4 translocation in skeletal muscle has the largest quantitative impact on whole-body glucose homeostasis. This is the cellular endpoint that chromium ultimately targets.
In chromium-deficient or insulin-resistant cells, GLUT4 vesicle trafficking is impaired at several steps: reduced AS160 phosphorylation (because upstream Akt activation is weaker), reduced vesicle docking at the plasma membrane (because downstream Rab proteins are inadequately activated), and reduced membrane fusion. The net effect is that fewer GLUT4 transporters reach the cell surface for a given insulin stimulus, and glucose accumulates in the bloodstream rather than entering muscle for storage as glycogen or oxidation for ATP.
Chromium supplementation, by amplifying the upstream insulin signal at the receptor level, propagates restoration of normal GLUT4 trafficking through the entire downstream cascade. Some long-term supplementation studies also suggest modest upregulation of GLUT4 protein content itself (via PGC-1-alpha-mediated transcriptional effects), which would constitute a second-order benefit beyond acute translocation enhancement.
Exercise increases GLUT4 translocation through an insulin-independent pathway (AMPK-mediated). This is one reason chromium supplementation has been studied as an adjunct in athletes and resistance trainers — the two interventions act on complementary pathways to maximize muscle glucose uptake during and after exercise. Lukaski's 1996 chromium picolinate plus resistance training trial in men is the classic citation here; results were modest but consistent with the expected directional effect.
Transferrin-Mediated Chromium Delivery
Chromium absorbed from the diet circulates in the bloodstream bound primarily to transferrin, the iron-transport protein. Each transferrin molecule has two metal-binding sites and can carry one or two chromium atoms in addition to or in place of iron. This shared transport mechanism has several consequences worth understanding:
- Iron overload competes with chromium delivery — in conditions of iron overload (hereditary hemochromatosis, repeated blood transfusion), transferrin sites are saturated with iron and chromium transport is impaired. Hemochromatosis patients can develop chromium deficiency despite normal dietary intake.
- Iron deficiency may enhance chromium delivery — the converse is biochemically plausible but clinically underdocumented. Anemic populations may have unusually high transferrin chromium-carrying capacity, though no clear clinical benefit has been mapped to this.
- Insulin signaling triggers chromium delivery — when insulin signaling activates a cell, the cell increases transferrin receptor expression on its surface, accelerating receptor-mediated endocytosis of transferrin-chromium complexes. Chromium is then released from transferrin in the endosome (similar to the iron-release mechanism) and exported to the cytoplasm where it loads onto apo-chromodulin.
- This is why chromium is specifically delivered to insulin-responsive tissues — non-insulin-target tissues do not upregulate transferrin receptor expression in response to insulin, so they receive proportionally less chromium. The mechanism is functionally elegant: chromium ends up where it is needed because the cells that need it actively recruit it.
This transferrin-chromium-insulin axis also explains why iron overload conditions (hemochromatosis, beta-thalassemia, transfusion-dependent anemia) sometimes present with mild glucose intolerance — impaired chromium delivery to insulin-target tissues plus direct iron-induced beta-cell toxicity both contribute. Iron-overloaded diabetic patients are an under-studied population in which chromium supplementation might have outsized benefit, though the clinical literature on this specific intersection remains sparse.
Non-Diabetic Insulin-Resistant Populations (PCOS, Prediabetes)
Insulin resistance is not exclusive to type 2 diabetes — it underlies several conditions where chromium has been studied as an intervention. The clinical evidence in non-diabetic populations is less robust than in established diabetes, but it is increasingly suggestive in two areas:
- Polycystic ovary syndrome (PCOS) — approximately 60–80% of women with PCOS have insulin resistance, and the resulting compensatory hyperinsulinemia drives ovarian androgen production (the proximate cause of the hirsutism, acne, and irregular menses that characterize the syndrome). Several small randomized trials of chromium picolinate (200–1,000 mcg/day for 8–24 weeks) have reported improvements in fasting insulin, HOMA-IR, and in some studies free testosterone and SHBG. The 2017 meta-analysis by Heshmati and colleagues (pooling six trials) found a statistically significant reduction in fasting insulin (-1.50 µIU/mL) and HOMA-IR (-0.38) with chromium picolinate in PCOS patients. The clinical magnitude is modest but consistent. Chromium is not a substitute for metformin in PCOS — metformin's effect on insulin sensitivity and ovulation is several-fold larger — but it is a reasonable adjunct for patients who tolerate metformin poorly or who prefer to start with nutritional approaches.
- Prediabetes — intermediate hyperglycemia (impaired fasting glucose 100–125 mg/dL, or HbA1c 5.7–6.4%) marks the window where intervention can prevent or substantially delay progression to type 2 diabetes. The largest evidence in this population comes from secondary analyses of mixed prediabetic-diabetic cohorts; dedicated prediabetes-only trials are relatively few. Effect sizes are smaller than in established diabetes, consistent with the general principle that insulin-sensitization benefits track with baseline impairment. Lifestyle modification (the Diabetes Prevention Program protocol of weight loss plus exercise) has a far larger effect on prediabetes-to-diabetes progression than any nutritional supplement; chromium is at best a small adjunct.
- Atypical depression with carbohydrate craving — covered in detail on the Weight Management deep-dive. Davidson 2003 RCT of 600 mcg/day chromium picolinate vs placebo in atypical depression showed significant improvement in carbohydrate craving and increased appetite scores, hypothesized to reflect serotonergic and noradrenergic effects downstream of improved insulin signaling in the central nervous system.
- Athletes and resistance trainers — aerobic exercise increases urinary chromium loss substantially. Chronic high-volume training in chromium-marginal individuals could theoretically produce mild deficiency. Practical impact in well-nourished athletes is small; the appropriate intervention is dietary diversification rather than routine supplementation.
Cautions and the Skeptical View
The skeptical position on chromium — well represented in the academic literature — deserves a fair hearing. The case for skepticism rests on several legitimate concerns:
- Heterogeneity of clinical trial results — not every trial replicates the Beijing 1997 effect size. Some well-conducted RCTs in Western diabetic populations have found essentially no benefit. The Balk meta-analysis aggregates this heterogeneity and finds a positive but modest pooled effect; a skeptic could reasonably argue that the modest effect reflects publication bias more than a true biological signal.
- No established deficiency biomarker — unlike vitamin D (serum 25-OH-D), B12 (serum methylmalonic acid), iron (ferritin), or zinc (serum zinc with caveats), there is no reliable serum or urinary marker for chromium status. Serum chromium is a poor marker of body stores; hair chromium reflects external contamination as much as internal status; urinary chromium reflects recent intake rather than reserves. This means we cannot easily identify the chromium-deficient subset of patients who would most benefit, and we cannot monitor response to supplementation biochemically.
- Mechanism is incompletely characterized — despite decades of work, the chromodulin story has not been fully nailed down at the structural-biology level. Crystal structures of chromodulin bound to the insulin receptor kinase domain do not exist (because chromodulin's exact structure remains debated). Skeptics argue that the eight-fold receptor amplification observed in vitro may not reflect physiologic intracellular chromium concentrations.
- NIH official position is cautious — the NIH Office of Dietary Supplements fact sheet on chromium concludes that "evidence is mixed" and that chromium supplementation cannot be recommended as a primary intervention for diabetes management. The European Food Safety Authority (EFSA) in 2014 went further and concluded that chromium is "not an essential element" because it could not identify a specific biochemical function that would be lost in the absence of dietary chromium — a controversial position that contradicts the TPN deficiency case reports.
- Practical caution: drug interactions — chromium can potentiate the glucose-lowering effect of insulin and oral antidiabetic drugs. Patients on these medications who start chromium without telling their physician risk hypoglycemia.
- Renal caution at very high doses — isolated case reports of acute renal injury at doses exceeding 1,200 mcg/day exist. Standard therapeutic doses (200–1,000 mcg/day) appear safe long-term in monitored populations.
- Pregnancy — chromium has been used in gestational diabetes trials without obvious harm, but the evidence base is too small for confident safety claims in pregnancy. Pregnant women should not start chromium supplementation without obstetric input.
The fair synthesis: chromium picolinate at 200–1,000 mcg/day is a reasonable adjunct for poorly controlled type 2 diabetes, PCOS with documented insulin resistance, and metabolic syndrome — with realistic expectations of modest rather than dramatic benefit. It is not a substitute for first-line interventions (lifestyle change, metformin, insulin where indicated). It is not indicated for non-diabetic adults with normal glucose tolerance. The skeptical position has merit but does not, in the author's view, justify dismissing a low-cost, low-toxicity intervention with a coherent mechanism and a positive (if heterogeneous) RCT literature.
Key Research Papers
- Anderson RA, Cheng N, Bryden NA, Polansky MM, Cheng N, Chi J, Feng J (1997). Elevated intakes of supplemental chromium improve glucose and insulin variables in individuals with type 2 diabetes. Diabetes 46(11):1786-1791. — PubMed
- Vincent JB (2000). The biochemistry of chromium. Journal of Nutrition 130(4):715-718. — PubMed
- Yamamoto A, Wada O, Suzuki H (1988). Purification and properties of biologically active chromium complex from bovine colostrum. Journal of Nutrition 118(1):39-45. — PubMed
- Davis CM, Vincent JB (1997). Chromium oligopeptide activates insulin receptor tyrosine kinase activity. Biochemistry 36(15):4382-4385. — PubMed
- Balk EM, Tatsioni A, Lichtenstein AH, Lau J, Pittas AG (2007). Effect of chromium supplementation on glucose metabolism and lipids: a systematic review of randomized controlled trials. Diabetes Care 30(8):2154-2163. — DOI: 10.2337/dc06-0996
- Heshmati J, Omani-Samani R, Vesali S, et al. (2018). The effects of supplementation with chromium on insulin resistance indices in women with polycystic ovarian syndrome: a systematic review and meta-analysis of randomized clinical trials. Hormone and Metabolic Research 50(03):193-200. — PubMed
- Mertz W (1993). Chromium in human nutrition: a review. Journal of Nutrition 123(4):626-633. — PubMed
- Vincent JB (2017). New evidence against chromium as an essential trace element. Journal of Nutrition 147(12):2212-2219. — PubMed
- EFSA NDA Panel (2014). Scientific opinion on dietary reference values for chromium. EFSA Journal 12(10):3845. — PubMed
- Lukaski HC, Bolonchuk WW, Siders WA, Milne DB (1996). Chromium supplementation and resistance training: effects on body composition, strength, and trace element status of men. American Journal of Clinical Nutrition 63(6):954-965. — PubMed
- Anderson RA, Polansky MM, Bryden NA, Patterson KY, Veillon C, Glinsmann WH (1983). Effects of chromium supplementation on urinary Cr excretion of human subjects and correlation of Cr excretion with selected clinical parameters. Journal of Nutrition 113(2):276-281. — PubMed
- Suksomboon N, Poolsup N, Yuwanakorn A (2014). Systematic review and meta-analysis of the efficacy and safety of chromium supplementation in diabetes. Journal of Clinical Pharmacy and Therapeutics 39(3):292-306. — PubMed
PubMed Topic Searches
- PubMed: Chromodulin and insulin receptor
- PubMed: Chromium picolinate and HOMA-IR
- PubMed: Chromium and GLUT4 translocation
- PubMed: Chromium and PCOS
- PubMed: Chromium transferrin transport
Connections
- Chromium Overview
- Chromium Benefits Hub
- Chromium for Blood Sugar
- Chromium for Lipid Profile
- Chromium for Weight Management
- Insulin Resistance
- Diabetes (Type 2)
- Metabolic Syndrome
- PCOS
- Fasting Insulin
- HbA1c
- Berberine
- Magnesium
- Iron (Transferrin Sharing)
- Vitamin B3 / Niacin
- Cinnamon
- Fasting
- Blood Sugar