Calcium and Muscle Function: Excitation-Contraction Coupling, Cramps, and the Mineral Balance
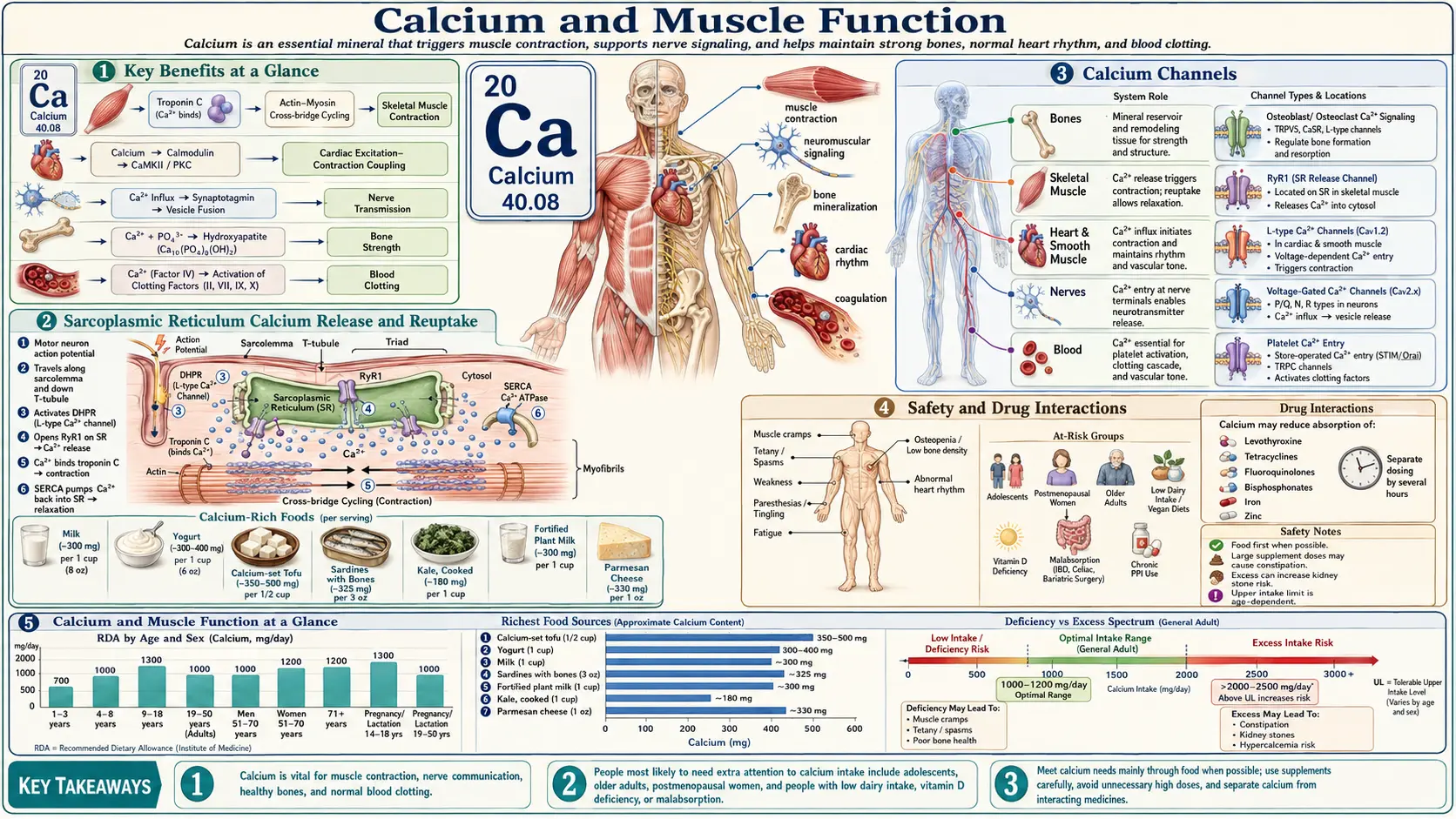
Calcium is the intracellular currency of muscle. Every heartbeat, every breath, every step depends on a precisely-timed release of Ca2+ from internal stores, its binding to a regulatory switch on the contractile filaments, and its rapid removal so the muscle can relax. The resting intracellular calcium concentration in a relaxed muscle fiber sits around 100 nM — roughly 10,000 times lower than the extracellular fluid bathing it. When the trigger fires, intracellular Ca2+ can rise to micromolar levels in milliseconds, drive force production, and then fall again before the next cycle. This page walks through that machinery in skeletal, cardiac, and smooth muscle; explains why deficient or excess intracellular calcium produces cramps, tetany, arrhythmia, and vasoconstriction; and reviews the real evidence for using dietary calcium and supplements to prevent muscle cramps.
Table of Contents
- Calcium as a Universal Signaling Ion
- Skeletal Muscle Excitation-Contraction Coupling
- Cardiac Muscle and Calcium-Induced Calcium Release
- Smooth Muscle and Vasoconstriction
- Deficiency Symptoms: Cramps, Tetany, Arrhythmia
- The Calcium-Magnesium Balance
- Vitamin D, Vitamin K2, and Potassium Interactions
- Dietary Calcium vs Supplement Forms
- Evidence for Cramp Prevention
- Hypercalcemia and Excess Risks
- Practical Recommendations
- Research Papers and References
- Connections
- Featured Videos
Calcium as a Universal Signaling Ion
Calcium is not used as a structural ion inside muscle cells — it is used as a switch. The cell maintains a steep electrochemical gradient by actively pumping Ca2+ out of the cytoplasm into either the extracellular space (via plasma-membrane Ca2+-ATPase, PMCA, and the Na+/Ca2+ exchanger, NCX) or into the sarcoplasmic reticulum (via SERCA, the sarco/endoplasmic reticulum Ca2+-ATPase). This gradient is the energy reservoir that makes contraction possible.
- Resting cytoplasmic [Ca2+] — approximately 100 nM (nanomolar).
- Activated cytoplasmic [Ca2+] — rises to 1–10 µM during contraction.
- SR luminal [Ca2+] — roughly 0.3–1 mM, buffered by calsequestrin.
- Extracellular [Ca2+] — approximately 1.2 mM ionized, of which serum protein binding leaves about 50% biologically active.
The fundamental insight is that contraction does not require calcium to be plentiful — it requires the gradient to be steep so that opening a small number of channels produces a fast, large rise. This is why blood calcium can vary 10-fold without affecting muscle force, but a sudden change in intracellular Ca2+ handling (a leaky ryanodine receptor, a slow SERCA pump) immediately changes contraction. The classic biochemical review of calcium as a signaling messenger remains Search PubMed.
Skeletal Muscle Excitation-Contraction Coupling
In skeletal muscle, the link between the membrane action potential and Ca2+ release is mechanical rather than chemical. The voltage-sensing dihydropyridine receptor (DHPR, the L-type Ca2+ channel CaV1.1) in the transverse-tubule membrane is physically coupled to the ryanodine receptor type 1 (RyR1) on the sarcoplasmic reticulum. The classic mechanism was worked out by Rios and Brum and synthesised in Search PubMed.
The full sequence:
- A motor neuron fires; acetylcholine is released at the neuromuscular junction.
- The endplate potential triggers an action potential that propagates along the sarcolemma and down the T-tubules.
- Depolarization moves the voltage-sensing S4 helices of DHPR. The mechanical conformational change is transmitted to RyR1.
- RyR1 opens. Ca2+ floods the cytosol from the terminal cisternae of the SR.
- Cytosolic Ca2+ binds the C-terminal lobe of troponin C (TnC).
- The conformational change in TnC pulls troponin I off actin and rotates tropomyosin out of the myosin-binding groove.
- Myosin heads (already cocked by ATP hydrolysis) bind exposed actin sites, perform the power stroke, release ADP, bind a fresh ATP, and detach — the cross-bridge cycle.
- SERCA1 pumps Ca2+ back into the SR (consuming 1 ATP per 2 Ca2+). Cytosolic Ca2+ falls, troponin releases, tropomyosin re-blocks actin, and the muscle relaxes.
The whole cycle — release, contraction, reuptake — can complete in under 50 ms in a fast twitch fiber. Comprehensive reviews of the molecular components include Search PubMed, and the more recent integrative model in Search PubMed.
Cardiac Muscle and Calcium-Induced Calcium Release
Cardiac muscle uses a different, calcium-dependent triggering mechanism. Here, the DHPR (CaV1.2 in heart) is a genuine Ca2+ channel rather than just a voltage sensor. The sequence is:
- Action potential depolarizes the cardiomyocyte sarcolemma.
- CaV1.2 opens during the plateau (phase 2). A small amount of extracellular Ca2+ enters the dyadic cleft.
- This trigger Ca2+ binds RyR2 on the SR, which opens to release a much larger pulse of Ca2+ from the SR — the calcium-induced calcium release (CICR) mechanism.
- Ca2+ binds cardiac TnC, contraction proceeds.
- SERCA2a (regulated by phospholamban) pumps Ca2+ back into the SR; the NCX extrudes Ca2+ from the cell in exchange for Na+.
The amount of trigger Ca2+ is graded, so the size of the resulting release is graded — this is why beta-adrenergic stimulation increases inotropy: cAMP-driven phosphorylation of CaV1.2, RyR2, phospholamban, and TnI all amplify and accelerate the Ca2+ transient. The definitive monograph-style review is Bers (2002), Nature, “Cardiac excitation-contraction coupling,” PMID 11805843 (doi:10.1038/415198a).
Cardiac arrhythmias are intimately tied to disordered Ca2+ handling. Leaky RyR2 channels (as in catecholaminergic polymorphic VT, or in heart failure) allow diastolic Ca2+ sparks that activate NCX, producing inward currents and triggered activity. Phospholamban mutations that disinhibit SERCA, or SERCA2a underexpression in failing myocardium, slow Ca2+ reuptake and impair both relaxation and the next contraction.
Smooth Muscle and Vasoconstriction
Smooth muscle lacks troponin entirely. Instead, the contractile switch is on the thick filament: myosin light-chain kinase (MLCK) phosphorylates the regulatory light chain of myosin, which then can interact with actin.
- A receptor agonist (norepinephrine on α1, angiotensin II on AT1, vasopressin on V1, etc.) activates Gq, which activates PLCβ.
- PLCβ cleaves PIP2 into IP3 and DAG.
- IP3 binds IP3R on the SR, releasing Ca2+. Concurrently, depolarization or store depletion opens L-type and store-operated (Orai/STIM) channels at the membrane.
- Ca2+ binds calmodulin (CaM), not troponin.
- Ca2+-CaM activates MLCK, which phosphorylates myosin RLC.
- Cross-bridges cycle; the vessel constricts.
- Relaxation occurs when Ca2+ is sequestered back into the SR or extruded by PMCA/NCX, and myosin light-chain phosphatase dephosphorylates RLC.
The pharmacology is clinically familiar: L-type Ca2+ channel blockers (amlodipine, nifedipine, verapamil, diltiazem) act predominantly on vascular smooth muscle, lowering peripheral resistance by reducing Ca2+ entry. The dihydropyridines (amlodipine, nifedipine) are vessel-selective; the non-dihydropyridines (verapamil, diltiazem) also slow AV-nodal conduction because they hit cardiac CaV1.2. A useful summary of smooth-muscle Ca2+ handling and its therapeutic exploitation is Search PubMed.
Deficiency Symptoms: Cramps, Tetany, Arrhythmia
The clinical picture of low ionized calcium (hypocalcemia, ionized Ca2+ below approximately 1.1 mmol/L) is dominated by neuromuscular hyperexcitability. The reason is paradoxical: extracellular Ca2+ normally stabilizes the resting membrane potential of nerves and muscle by neutralizing surface negative charges. Lowering extracellular Ca2+ increases membrane excitability — nerves fire on their own, muscles twitch spontaneously, and sustained involuntary contractions appear.
- Paresthesias — perioral and finger-tip tingling are the earliest symptoms.
- Carpopedal spasm — flexion of the wrist and metacarpophalangeal joints with extension of the interphalangeal joints (the “obstetrician’s hand”).
- Chvostek’s sign — tapping the facial nerve in front of the ear produces ipsilateral facial twitching.
- Trousseau’s sign — inflating a sphygmomanometer cuff above systolic pressure for 3 minutes induces carpal spasm.
- Tetany — generalized sustained muscle contraction.
- Laryngospasm and bronchospasm — can compromise the airway.
- Seizures — from cortical hyperexcitability.
- Prolonged QT interval — the cardiac plateau is calcium-dependent; hypocalcemia lengthens the ST segment and predisposes to torsades de pointes.
- Heart failure — chronic, severe hypocalcemia reduces cardiac contractility and can present as reversible cardiomyopathy.
Most patients with cramps do not have systemic hypocalcemia. True hypocalcemic tetany is a hospital problem (hypoparathyroidism after thyroidectomy, vitamin D deficiency, magnesium depletion preventing PTH release, citrate from massive transfusion, severe pancreatitis). Ordinary nocturnal leg cramps and exercise-associated muscle cramps almost always occur with normal serum calcium. A useful general clinical review is Search PubMed.
The Calcium-Magnesium Balance
Calcium and magnesium are physiological partners and competitors at almost every step of muscle function. Magnesium acts as a natural calcium antagonist: it competes for binding sites on troponin C (with lower affinity), on calmodulin, on RyR (which it inhibits), and on L-type calcium channels (which it blocks at high concentration). Functionally, calcium drives contraction; magnesium permits relaxation. The classic biophysical work is summarized in Search PubMed.
Three practical consequences:
- Magnesium deficiency mimics excess calcium signaling. Low intracellular Mg2+ leaves RyR more leaky and reduces SERCA efficiency — cytosolic Ca2+ stays elevated for longer, the muscle relaxes incompletely, and cramps result.
- Hypomagnesemia causes functional hypocalcemia. Magnesium is required for both PTH secretion and PTH receptor responsiveness. A patient with persistent “refractory” hypocalcemia is usually magnesium-depleted; the calcium will not correct until the magnesium does.
- Loading calcium without addressing magnesium can worsen cramps. Pushing dietary or supplemental calcium without correcting an underlying magnesium deficit can paradoxically increase muscle excitability. The aim is balance, not maximizing one mineral.
The mechanistic relationship between Mg2+ and the cardiovascular system, including Mg2+-driven Ca2+ channel block, is reviewed in de Baaij, Hoenderop & Bindels (2015), Physiol Rev, PMID 25540137 (doi:10.1152/physrev.00012.2014).
Vitamin D, Vitamin K2, and Potassium Interactions
Calcium does not work alone. Three companion nutrients control where calcium goes and what it does:
- Vitamin D3 (calcitriol). The active hormone 1,25-(OH)2-D3 drives intestinal Ca2+ absorption (TRPV6, calbindin, PMCA1b) and renal reabsorption. Without adequate vitamin D, dietary calcium intake matters far less because little is absorbed. Severe deficiency presents as rickets in children and osteomalacia in adults, both of which produce muscle weakness and bone pain. Vitamin D also has direct effects on muscle — receptors are present in skeletal muscle, and deficiency is associated with proximal weakness and falls. See Search PubMed.
- Vitamin K2 (menaquinones). K2 is the cofactor for γ-carboxylation of two key Gla proteins: osteocalcin (which binds Ca2+ into bone) and matrix Gla protein (MGP, which keeps Ca2+ out of arteries). Inadequate K2 leaves osteocalcin uncarboxylated (Ca2+ is poorly deposited in bone) and MGP uncarboxylated (Ca2+ deposits in arteries instead). The randomized trial showing K2 (MK-7) reduces arterial stiffness and slows bone loss is Search PubMed.
- Potassium. Potassium sets the resting membrane potential; sodium drives the depolarization; calcium triggers contraction. Hypokalemia depolarizes resting potential, inactivates Na+ channels, and causes weakness and cramps. Hyperkalemia depolarizes membranes towards threshold initially (irritable) and then inactivates them (flaccid). Aldosterone-driven potassium loss from diuretics or licorice abuse is a classic correctable cause of muscle cramping; the calcium-potassium pair should be considered together.
Dietary Calcium vs Supplement Forms
Adult calcium needs are approximately 1,000 mg/day (1,200 mg over age 50 in women, over age 70 in men) per the Institute of Medicine. Most of this is best obtained from food, where calcium comes packaged with a food matrix that controls the rate of absorption.
- Dairy — 250–300 mg per cup of milk or yogurt; bioavailability about 30%, helped by lactose and casein phosphopeptides.
- Sardines and canned salmon with bones — 300–325 mg per 3 oz serving; also delivers vitamin D, omega-3s, and protein.
- Tofu set with calcium sulfate — 250–350 mg per ½ cup.
- Leafy greens with low oxalate — bok choy, kale, collards, mustard greens (50–90 mg per cup cooked, with 50–60% bioavailability — higher than dairy on a per-mg basis).
- Spinach, Swiss chard, beet greens — high calcium but high oxalate; absorbed fraction is only about 5%.
- Fortified plant milks and juices — 300 mg per cup when fortified with calcium carbonate or tricalcium phosphate.
- Almonds and sesame seeds (tahini) — modest contributors.
If supplementation is needed, the form matters:
- Calcium carbonate — 40% elemental calcium by weight (the highest of any common form). Cheap. Requires gastric acid; must be taken with food. Poorly absorbed in achlorhydria or on PPIs. Common GI side effects: constipation, gas.
- Calcium citrate — 21% elemental. Absorbed independent of gastric acid; good choice for older adults, those on acid blockers, and those with low stomach acid. Higher pill burden because elemental fraction is lower.
- Calcium citrate-malate — the most bioavailable common form; used in many fortified juices.
- Calcium hydroxyapatite (microcrystalline / MCHA) — whole-bone derivative; provides calcium with phosphorus and trace bone-matrix proteins. Evidence is mixed; quality control varies.
- Calcium lactate, gluconate — low elemental fraction (13% and 9%); used in IV preparations and food fortification.
Cap individual doses at 500–600 mg elemental calcium; absorption fraction falls steeply above that. Take with vitamin D3 (1,000–2,000 IU) and ideally menaquinone-7 (90–180 µg) to direct deposition to bone rather than arteries.
Evidence for Cramp Prevention
The honest answer is that the evidence base for calcium specifically as a cramp preventer is weak, because most cramps are not driven by hypocalcemia. The most-studied scenario is pregnancy-associated leg cramps, where the Cochrane review found magnesium supplementation showed inconsistent benefit, calcium showed little benefit, and vitamin B-complex showed some benefit: Search PubMed.
For exercise-associated muscle cramps, the consensus has shifted away from the “electrolyte depletion” theory toward a neuromuscular control hypothesis — fatigued motor neurons firing aberrantly. Supplementing calcium, sodium, or magnesium produces little measurable benefit in randomized trials; pickle juice (with its acetic acid trigger of oropharyngeal TRPV1/A1 receptors and presumed inhibition of alpha-motor neurons) has been more compelling than any electrolyte. See Search PubMed.
For idiopathic nocturnal leg cramps in older adults, the systematic review of treatments found that quinine has the best (still modest) evidence, calcium supplementation is no better than placebo, and magnesium results are inconsistent: Search PubMed. The practical reality is that for any patient with persistent cramps, the workup is to check magnesium, potassium, ionized calcium, vitamin D, and TSH — correct what is low — rather than assume calcium is the answer.
Hypercalcemia and Excess Risks
Too much intracellular or extracellular calcium is just as dangerous as too little. Hypercalcemia (serum total Ca above approximately 10.5 mg/dL) initially causes mild symptoms (“stones, bones, groans, psychic moans” — nephrolithiasis, bone pain, abdominal discomfort, fatigue, depression) and at higher levels causes confusion, weakness, dehydration, and arrhythmias including shortened QT and AV blocks.
- Primary hyperparathyroidism — the leading outpatient cause; usually a single adenoma.
- Malignancy — PTHrP-secreting tumors, lytic bone metastases, multiple myeloma; the leading inpatient cause.
- Vitamin D toxicity — rare but possible at sustained intakes above 10,000 IU/day or in granulomatous disease (sarcoidosis) with extra-renal 1α-hydroxylase activity.
- Milk-alkali syndrome — high calcium intake (especially calcium carbonate antacids) with alkaline load.
- Thiazide diuretics — reduce urinary calcium excretion.
Beyond systemic hypercalcemia, the question of supplemental calcium and cardiovascular risk has been hotly debated. A meta-analysis showed supplemental calcium without vitamin D was associated with a small increase in myocardial infarction; pooled trial data have been inconsistent: Bolland et al. (2010), BMJ, PMID 20671013 (doi:10.1136/bmj.c3691). The likely mechanism is that bolus supplementation transiently raises serum calcium and, in the absence of K2-driven MGP carboxylation, may accelerate vascular calcification. Pairing calcium with vitamin K2 (and obtaining calcium from food when possible) appears to mitigate this concern.
At the cellular level, excess intracellular calcium is the final common pathway for many forms of cell death. Mitochondrial Ca2+ overload opens the permeability transition pore, releasing cytochrome c and triggering apoptosis — central to ischemia-reperfusion injury, excitotoxic neuronal death, and rhabdomyolysis. Calpains activated by sustained Ca2+ elevation digest cytoskeleton and contractile proteins; this is part of why severe, prolonged muscle ischemia destroys the fiber even after blood flow returns.
Practical Recommendations
- Aim for 1,000–1,200 mg total daily calcium from diet first. Dairy, sardines, low-oxalate greens, and fortified foods make this achievable without supplements for most people.
- If supplementing, choose citrate over carbonate if you are over 60, on PPIs/H2 blockers, or have any reason to suspect low stomach acid. Take with meals. Cap individual doses at 500–600 mg elemental.
- Always pair calcium with vitamin D3 (1,000–2,000 IU/day; check a 25-OH-D level and aim for 40–60 ng/mL).
- Add vitamin K2 (MK-7, 90–180 µg/day) if you are taking any calcium supplementation, especially if you have cardiovascular risk factors. Skip if you are on warfarin without consulting your prescriber.
- Address magnesium first if you have cramps. Most cramping is more responsive to magnesium glycinate or magnesium citrate (200–400 mg elemental/day) than to additional calcium. See Magnesium.
- Check potassium if you are on a diuretic, have GI losses, or use licorice or excessive caffeine.
- Investigate persistent cramps with magnesium, potassium, ionized calcium, 25-OH vitamin D, TSH, and a careful medication review (statins, diuretics, beta-agonists are common offenders).
- For exercise cramps, look beyond electrolytes: pacing, conditioning, stretching, and rapid neuromuscular “reset” (pickle juice, mustard) are often more effective than electrolyte loading.
- For cardiac concerns, get calcium from food rather than supplements when possible; if supplementing, pair with vitamin K2 to direct calcium to bone.
Research Papers and References
- Search PubMed
- Search PubMed
- Search PubMed
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415(6868):198-205. PMID 11805843. doi:10.1038/415198a
- Search PubMed
- Search PubMed
- Search PubMed
- de Baaij JHF, Hoenderop JGJ, Bindels RJM. Magnesium in man: implications for health and disease. Physiol Rev. 2015;95(1):1-46. PMID 25540137. doi:10.1152/physrev.00012.2014
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ. 2010;341:c3691. PMID 20671013. doi:10.1136/bmj.c3691
Connections
- All Minerals
- Calcium Overview
- Calcium for Bone Health
- Calcium for Cardiovascular Health
- Calcium for Muscle Function (Benefits sub-article)
- Calcium for Nerve Transmission
- Magnesium
- Potassium
- Phosphorus
- Vitamin D3
- Vitamin K (K2/MK-7)
- Cramp Prevention
- Hypertension and Calcium Channel Blockers
- Osteoporosis
- Milk
- Collagen