Pau d'Arco — Cautions & Cancer Research
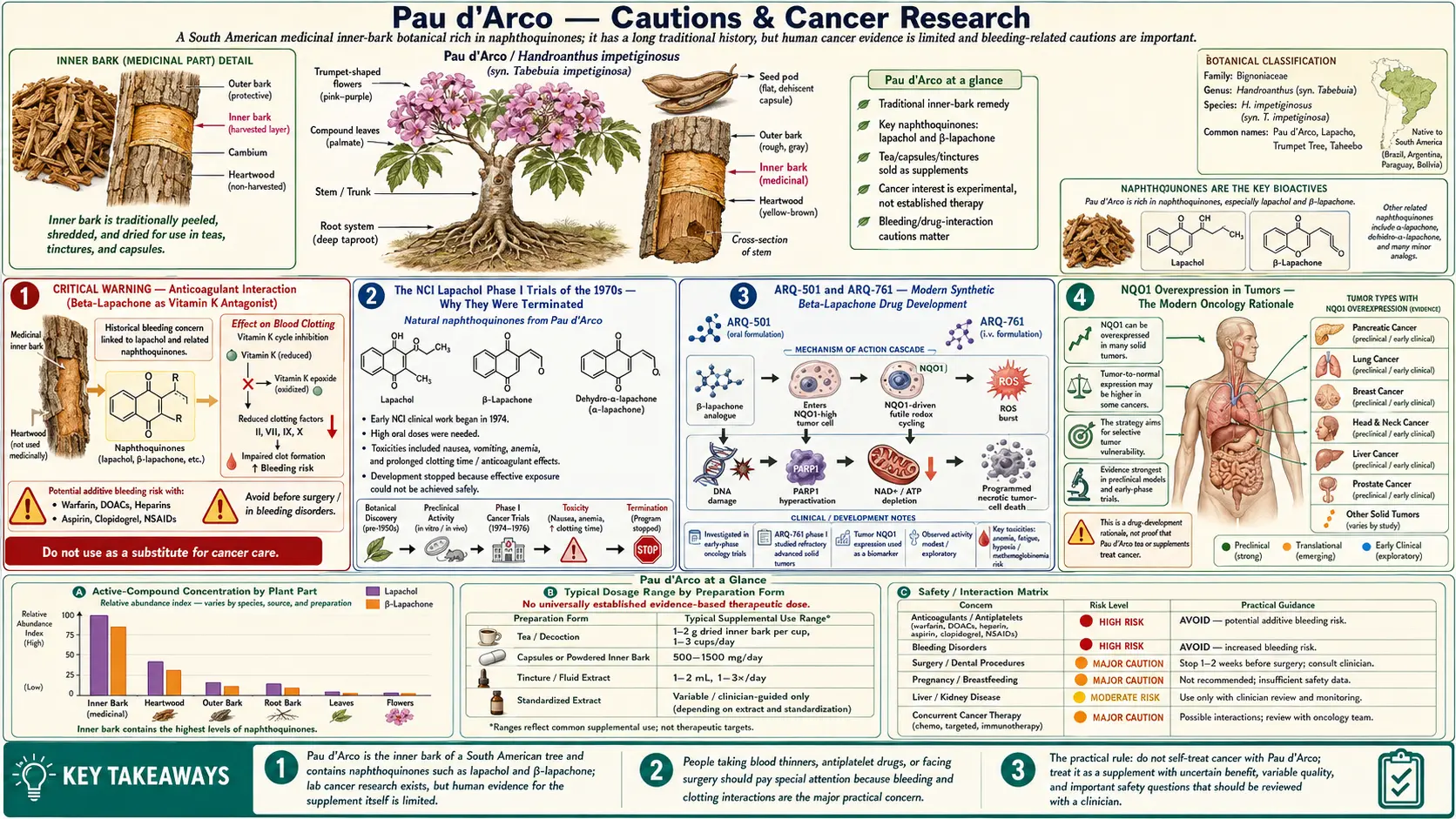
Pau d'Arco occupies an unusual place in the history of cancer research: in the 1970s the National Cancer Institute (NCI) ran Phase I clinical trials of lapachol as an antitumor agent, and the trials were terminated due to dose-limiting toxicity — prothrombin time prolongation, nausea, and vomiting at the doses required for antitumor effect. The story did not end there. Synthetic beta-lapachone derivatives (most prominently ARQ-501, later ARQ-761) entered modern oncology Phase I/II trials in the early 2000s as topoisomerase I/II poisons, and active research continues. For the patient, the relevant lessons are practical: there is a real anticoagulant interaction (do not combine with warfarin, heparin, DOACs, or antiplatelet drugs), a real high-dose toxicity profile that limits safe oral dosing, an absolute pregnancy contraindication, and several specific drug interactions that need awareness. This page is the safety reference for every other Pau d'Arco page on the site — read it before any practical use.
Table of Contents
- CRITICAL WARNING — Anticoagulant Interaction (Beta-Lapachone as Vitamin K Antagonist)
- The NCI Lapachol Phase I Trials of the 1970s — Why They Were Terminated
- ARQ-501 and ARQ-761 — Modern Synthetic Beta-Lapachone Drug Development
- NQO1 Overexpression in Tumors — The Modern Oncology Rationale
- High-Dose Hepatotoxicity, Hematologic Toxicity, and Nephrotoxicity
- Pregnancy, Breastfeeding, and Teratogenicity — ABSOLUTE Contraindication
- G6PD Deficiency and Hemolytic Anemia Risk
- Drug Interactions Beyond Anticoagulants
- Absolute and Relative Contraindications — Quick Reference
- Monitoring Recommendations for Sustained Use
- Pau d'Arco and Cancer Patients — A Realistic Discussion
- Key Research Papers
- Connections
- Featured Videos
CRITICAL WARNING — Anticoagulant Interaction (Beta-Lapachone as Vitamin K Antagonist)
The single most consequential safety issue with Pau d'Arco is the vitamin K antagonist functional activity of beta-lapachone and lapachol. Both molecules share structural similarity to warfarin's 4-hydroxycoumarin pharmacophore and have documented prothrombin time prolongation in human and animal studies. The NCI Phase I lapachol trials in the 1970s were specifically terminated because of this effect — antitumor-effective doses of oral lapachol produced significant INR elevation in a substantial fraction of patients, with measurable risk of bleeding.
This means Pau d'Arco is contraindicated in combination with:
- Warfarin (Coumadin, Jantoven) — absolute contraindication. The mechanisms are synergistic. Concurrent use can produce dangerous INR elevation and bleeding.
- Direct oral anticoagulants (DOACs) — apixaban (Eliquis), rivaroxaban (Xarelto), edoxaban (Savaysa), dabigatran (Pradaxa). The bleeding risk is additive.
- Heparin and low-molecular-weight heparin (enoxaparin, dalteparin, fondaparinux) — additive bleeding risk.
- Antiplatelet drugs — clopidogrel (Plavix), ticagrelor (Brilinta), prasugrel (Effient), ticlopidine. Bleeding risk is additive.
- Aspirin at antiplatelet doses (75-325 mg/day) — bleeding risk is additive. The risk is more pronounced when aspirin is combined with another antiplatelet (dual antiplatelet therapy) or with an anticoagulant.
- Other NSAIDs at chronic doses — ibuprofen, naproxen, celecoxib, meloxicam, diclofenac. Lesser concern than the explicit anticoagulants but still measurable additive bleeding risk.
- Fish oil at high doses (greater than 3 g/day combined EPA+DHA) — mild platelet-inhibition effect that can be additive.
- Other herbal anticoagulants — high-dose garlic, ginkgo, ginger, dong quai, feverfew. Mild additive effects.
SURGICAL PRECAUTION: Discontinue Pau d'Arco at least 14 days before any planned surgical procedure, dental extraction, biopsy, or invasive imaging procedure. This includes elective cosmetic procedures. Notify all surgical and anesthesia teams in advance about Pau d'Arco use, even if recently discontinued. The same applies to any planned dental work involving deep cleaning or extraction.
EMERGENCY SITUATIONS: Patients taking Pau d'Arco who present to emergency departments with bleeding, head injury, or planned emergency surgery should explicitly disclose the use to the treating team. INR may be elevated even when not on warfarin.
The NCI Lapachol Phase I Trials of the 1970s — Why They Were Terminated
The National Cancer Institute screened thousands of plant compounds for antitumor activity during the 1950s-1970s. Lapachol (designated NSC-11905 in the NCI compound registry) was identified as showing reproducible activity against several human tumor cell lines and against the Walker 256 carcinosarcoma rat model. The initial preclinical data were impressive enough to justify Phase I clinical trials in humans, which began in the late 1960s.
The Phase I trial published by Block, Serpick, Miller, and Wiernik in 1974 (Cancer Chemotherapy Reports) is the definitive published account. Twenty-one patients with advanced cancer received escalating oral doses of lapachol, beginning at 250 mg/day and titrated upward. The findings:
- At doses below 1.5 g/day, no antitumor effect was observed (suggesting the threshold for tumor response was higher than the antifungal / anti-inflammatory dose range used in folk medicine)
- At doses of 1.5-3.5 g/day, modest antitumor effects were observed in some patients (partial tumor regression, decreased pain, improved performance status)
- Dose-limiting toxicities at these higher doses were:
- Prothrombin time prolongation with measurable bleeding risk — the dominant safety issue
- Severe nausea and vomiting (poorly controlled by 1970s antiemetics)
- Anemia in several patients
- The dose required for antitumor effect was at or above the dose producing unacceptable toxicity. The therapeutic index was too narrow to justify continued development of natural lapachol as an oncology drug.
The trials were formally closed. Subsequent NCI interest in the lapachol scaffold turned toward chemical modification to separate the antitumor activity from the prothrombin-prolonging effect — this is the work that ultimately led to the modern beta-lapachone derivative programs.
The clinical implication for natural Pau d'Arco supplementation: the doses used in modern integrative medicine for antifungal, immune-modulating, and anti-inflammatory effect (typically 500-1,500 mg of dried-bark extract twice daily) are well below the doses where the NCI saw significant bleeding risk in the Phase I trials. However, the prothrombin effect is dose-dependent rather than binary — lower doses still produce smaller effects on coagulation, which become clinically meaningful when combined with anticoagulant or antiplatelet drugs.
ARQ-501 and ARQ-761 — Modern Synthetic Beta-Lapachone Drug Development
ArQule, Inc. (a biotech later acquired by Merck) developed a series of synthetic beta-lapachone derivatives in the early 2000s as candidate oncology agents. The lead compound, ARQ-501 (beta-lapachone in a hydroxypropyl-beta-cyclodextrin formulation), entered NCI-sponsored Phase I trials around 2003 for solid tumors. A second-generation compound, ARQ-761, followed with improved pharmacokinetics.
The mechanism that distinguishes these synthetic derivatives from generic naphthoquinone toxicity is their selective activation by NAD(P)H:quinone oxidoreductase 1 (NQO1), an enzyme that is markedly overexpressed in many human tumors (particularly lung, breast, pancreatic, and head and neck cancers) but expressed at low levels in most normal tissues. NQO1 reduces beta-lapachone to an unstable hydroquinone that rapidly reoxidizes, depositing reactive oxygen species and triggering DNA strand breaks specifically in NQO1-high tumor cells. This selectivity is the key drug-development rationale.
Phase I and early Phase II trial results have been mixed. ARQ-501 showed acceptable safety in the cyclodextrin-formulated intravenous form, with some signals of activity, but did not achieve the response rates required for further development as monotherapy. ARQ-761 continued through Phase I in combination with chemotherapy. As of the most recent published trials, neither compound has achieved FDA approval, and the active development programs have been deprioritized.
The longer-term significance is twofold:
- The naphthoquinone-NQO1 selective-activation concept has been validated as biologically real and is being explored with newer chemical scaffolds
- The clinical experience with ARQ-501 and ARQ-761 confirmed the toxicity profile observed in the 1970s lapachol trials: methemoglobinemia, hemolytic anemia, prothrombin time prolongation, and dose-limiting nausea remain the principal challenges
None of this should be over-interpreted to suggest that natural Pau d'Arco supplementation is an effective cancer treatment. The synthetic derivative doses, intravenous formulation, and pharmacokinetic profile are fundamentally different from oral whole-bark extract supplementation, and the synthetic compounds have not achieved FDA approval even with the optimization of decades of medicinal chemistry effort.
NQO1 Overexpression in Tumors — The Modern Oncology Rationale
The mechanism behind selective tumor killing by beta-lapachone and its synthetic derivatives is worth understanding because it explains both the rational basis for the oncology drug-development programs and the realistic limits of what oral natural Pau d'Arco can achieve.
NQO1 (NAD(P)H:quinone oxidoreductase 1, also called DT-diaphorase) is a cytoplasmic flavoenzyme that normally functions as a phase-2 detoxification enzyme — it performs two-electron reduction of quinones to relatively stable hydroquinones, bypassing the redox-cycling semiquinone intermediate. For most substrates, NQO1 is protective.
For beta-lapachone, the situation is unusual: the two-electron reduction product is unstable and rapidly reoxidizes to the parent quinone, generating large amounts of superoxide and depleting NAD(P)H. The net effect is intracellular oxidative catastrophe specifically in cells with high NQO1 activity.
NQO1 is overexpressed 5-200-fold in many human tumors relative to adjacent normal tissue. This includes:
- Non-small cell lung cancer (NSCLC) — particularly the squamous subtype
- Pancreatic ductal adenocarcinoma
- Triple-negative breast cancer
- Head and neck squamous cell carcinoma
- Some colorectal and ovarian cancers
This tumor-selective expression is the foundation of the rational drug-development effort. The unfinished story is that achieving therapeutic intratumoral concentrations of beta-lapachone with acceptable systemic toxicity remains difficult, even with the synthetic derivatives.
High-Dose Hepatotoxicity, Hematologic Toxicity, and Nephrotoxicity
Beyond the anticoagulation concern, sustained high-dose oral Pau d'Arco can produce:
- Hepatotoxicity — ALT and AST elevation, occasionally cholestatic patterns, rarely hepatocellular injury. The mechanism is the same redox-cycling oxidative stress that drives the antifungal and antitumor effects, applied to a tissue (hepatocyte) that is metabolically active and exposed to first-pass concentration of orally absorbed naphthoquinones. Patients with pre-existing liver disease (chronic hepatitis B or C, fatty liver, alcoholic liver disease) should be especially cautious and should have baseline liver function tested.
- Hemolytic anemia — the oxidative stress on red blood cells can produce hemolysis, particularly in patients with G6PD deficiency or other red cell antioxidant defects. Symptoms include unusual fatigue, jaundice, dark urine, and decreased exercise tolerance.
- Methemoglobinemia — documented in the synthetic derivative trials (ARQ-501, ARQ-761) at high parenteral doses; theoretically possible but less commonly reported with oral whole-bark extract at lower doses. Symptoms: cyanosis disproportionate to oxygen saturation, headache, fatigue.
- Nephrotoxicity — mild creatinine elevation has been reported with sustained high-dose use. Mechanism likely a combination of direct oxidative stress on renal tubular cells and reduced renal perfusion from any bleeding effect. Patients with pre-existing chronic kidney disease should avoid high-dose Pau d'Arco.
- Gastrointestinal toxicity — nausea, vomiting, epigastric pain, diarrhea. Common at higher doses, generally tolerable at the integrative-medicine dose range when taken with meals.
- Bone marrow suppression — documented at the NCI Phase I antitumor doses; not generally seen at the integrative-medicine dose range. Patients on concurrent myelosuppressive chemotherapy should not add Pau d'Arco without explicit oncology team approval.
For courses longer than 8 weeks at the upper dose range, periodic laboratory monitoring (CBC, comprehensive metabolic panel including ALT/AST/total bilirubin/creatinine) is reasonable. Stop immediately for any unexplained jaundice, dark urine, fatigue, easy bruising, or persistent right-upper-quadrant discomfort.
Pregnancy, Breastfeeding, and Teratogenicity — ABSOLUTE Contraindication
Pau d'Arco is absolutely contraindicated in pregnancy. Lapachol crosses the placenta and has demonstrated reproductive and developmental toxicity in rodent studies. The specific findings have included:
- Increased fetal resorption (early pregnancy loss) in rats
- Skeletal malformations
- Decreased fetal weight
- Maternal hemorrhage at higher doses (compounding the vitamin K antagonist effect with the increased vascular fragility of pregnancy)
No formal teratogenicity studies in humans exist (and ethically cannot be conducted), so the assignment is by precaution. The combination of (a) animal teratogenicity, (b) anticoagulant activity in a setting where maternal hemorrhage is already a concern, (c) immune-modulating activity in a setting where pregnancy-appropriate immune tolerance is required, and (d) the availability of safer alternatives for every clinical indication, all support the absolute pregnancy contraindication.
The contraindication applies to all forms and all doses:
- Oral capsules, tinctures, decoctions, teas
- Topical use over large body-surface areas (small patches of topical use, e.g., for a localized fungal skin lesion the size of a coin, are likely safe but should still be discussed with maternal-fetal medicine)
- Vaginal suppositories — absolutely avoid in pregnancy
Breastfeeding is also a contraindication. Lapachol passes into breast milk and is biologically active. The neonatal liver is not fully mature with respect to the cytochrome P450 enzymes that detoxify naphthoquinones, and the neonatal antioxidant defenses are less robust. Safer alternatives exist for every indication.
Women trying to conceive should also avoid Pau d'Arco during the trying-to-conceive interval, since pregnancy may be confirmed several weeks after conception and the highest teratogenic risk is in the first 8-10 weeks of gestation.
G6PD Deficiency and Hemolytic Anemia Risk
Glucose-6-phosphate dehydrogenase (G6PD) is the first enzyme in the pentose phosphate pathway and is critical for generating NADPH, the cellular currency for reducing glutathione (GSH) and other antioxidant systems. G6PD deficiency — an X-linked inherited enzyme defect — affects approximately 400 million people worldwide, with the highest prevalence in Mediterranean, African, Middle Eastern, and Southeast Asian populations. Affected individuals have inadequate NADPH regeneration in red blood cells, making the red cells especially vulnerable to oxidative stress.
Pau d'Arco's redox-cycling naphthoquinones impose precisely this kind of oxidative stress. G6PD-deficient patients exposed to lapachol or beta-lapachone are at increased risk for:
- Acute hemolytic anemia — potentially severe, with abrupt drop in hemoglobin, jaundice, dark urine, and hemoglobinuria
- Heinz body formation on peripheral blood smear (oxidized hemoglobin denaturation)
- Methemoglobinemia
Patients of Mediterranean, African, Middle Eastern, Sephardic Jewish, Greek, Southern Italian, or Southeast Asian ancestry should consider G6PD testing before starting Pau d'Arco at any oral dose. The test is widely available and inexpensive.
Other commonly used substances also problematic in G6PD deficiency include sulfonamide antibiotics, dapsone, nitrofurantoin, primaquine, fava beans, and methylene blue. The list of "G6PD-deficient avoidance" agents is maintained by the American Society of Hematology and is worth reviewing for any patient with confirmed G6PD deficiency.
Drug Interactions Beyond Anticoagulants
In addition to the anticoagulant interactions covered above, Pau d'Arco has several other documented or theoretical drug interactions worth awareness:
- Topoisomerase inhibitor chemotherapy — irinotecan (Camptosar), topotecan (Hycamtin), etoposide (VePesid), teniposide, doxorubicin (Adriamycin), epirubicin, mitoxantrone. Beta-lapachone is itself a topoisomerase poison, so concurrent use has a theoretical mechanistic concern. Patients receiving topoisomerase-targeted chemotherapy should discuss any Pau d'Arco use with the treating oncology team.
- Hepatotoxic drugs — acetaminophen (paracetamol) at high doses, methotrexate, isoniazid, amiodarone, valproic acid, several statins. The additive hepatotoxicity risk warrants monitoring or avoidance of concurrent use at high doses.
- Other vitamin K antagonists — phenindione, acenocoumarol, fluindione (rarely used in U.S.). Same risk as warfarin.
- Immunosuppressants — cyclosporine, tacrolimus, mycophenolate, azathioprine, biologic DMARDs. Pau d'Arco's immune-modulating effects could theoretically interfere with the desired immunosuppression in organ transplant recipients. Avoid in this population without explicit transplant team approval.
- Calcineurin inhibitors specifically — cyclosporine and tacrolimus are metabolized by CYP3A4. Pau d'Arco effects on CYP enzymes are not well characterized, but a theoretical risk of altered drug levels exists.
- Hypoglycemic drugs — some animal studies suggest Pau d'Arco may have modest hypoglycemic effect. Diabetic patients on insulin or sulfonylurea should monitor glucose more frequently when starting Pau d'Arco.
- Diuretics and antihypertensives — no specific interaction documented, but the modest diuretic effect of some Pau d'Arco preparations could theoretically be additive.
- Other immune-stimulating herbs — echinacea, andrographis, astragalus, cat's claw. Additive immune modulation, generally well tolerated but worth awareness in patients with autoimmune disease.
Absolute and Relative Contraindications — Quick Reference
Absolute contraindications (do NOT use):
- Pregnancy at any stage
- Breastfeeding
- Concurrent warfarin, DOAC, heparin, or therapeutic-dose anticoagulation
- Active bleeding or recent significant bleeding (GI bleed, intracranial hemorrhage)
- Planned surgery within the next 14 days
- Known severe G6PD deficiency
- Active solid organ transplant on immunosuppression (unless transplant team explicitly approves)
- Pediatric use under age 6 (insufficient safety data)
Relative contraindications (use only with caution and physician involvement):
- Concurrent antiplatelet therapy (aspirin, clopidogrel)
- Chronic NSAID use
- Chronic liver disease (hepatitis B/C, advanced fatty liver, cirrhosis)
- Chronic kidney disease stage 3 or worse
- Active Th1-dominant autoimmune disease (Hashimoto's, MS, type 1 diabetes, RA)
- Pediatric use ages 6-12 (lower doses, shorter duration, with pediatrician awareness)
- Frail elderly with polypharmacy
- Active cancer chemotherapy without oncology team awareness
- Patients with bleeding disorders (hemophilia, von Willebrand disease, immune thrombocytopenia)
- History of hemolytic anemia from any cause
Monitoring Recommendations for Sustained Use
For patients taking Pau d'Arco at standard integrative-medicine doses (500-1,500 mg twice daily of standardized extract) for more than 8 weeks, basic laboratory monitoring is appropriate:
- Baseline (before starting): CBC with differential, comprehensive metabolic panel (CMP) including ALT, AST, total bilirubin, alkaline phosphatase, creatinine, BUN. PT/INR if there is any history of bleeding tendency or any concurrent antiplatelet/anticoagulant use.
- At 8 weeks of continuous use: Repeat CBC and CMP. Consider PT/INR if not done at baseline.
- Every 12 weeks thereafter for continuous use: CBC and CMP.
- Any time symptoms develop: New onset jaundice, unusual fatigue, easy bruising, blood in urine or stool, dark urine, persistent right-upper-quadrant pain — stop Pau d'Arco immediately and obtain CBC, CMP, PT/INR.
For courses of 8 weeks or less at standard doses in otherwise healthy patients, monitoring is generally not required — the safety experience supports use without laboratory burden in low-risk patients.
The practical recommendation for most patients is intermittent rather than continuous use: 8-12 weeks of active treatment, then a 2-4 week break, then repeat as needed. This pattern allows assessment of whether sustained use is still providing benefit, reduces the risk of cumulative toxicity, and provides natural opportunities to reassess the underlying clinical situation.
Pau d'Arco and Cancer Patients — A Realistic Discussion
Pau d'Arco has been promoted in some alternative-medicine circles as a "natural cancer cure" — a claim that does not align with the actual evidence. The history is worth understanding honestly:
- What the NCI trials showed: lapachol has real but modest antitumor activity in some patients at doses that produce dose-limiting toxicity. The therapeutic index is too narrow for clinical use as natural oral lapachol. This was the NCI's formal conclusion in the 1970s.
- What the synthetic-derivative programs have shown: ARQ-501 and ARQ-761 are biologically active in NQO1-high tumors, but have not achieved the response rates required for FDA approval even after years of medicinal chemistry optimization. Active research continues but no approved drug has emerged.
- What anecdotal reports show: Individual case reports of tumor stabilization or regression in patients self-treating with Pau d'Arco exist but cannot be interpreted as evidence of efficacy in the absence of controlled trials and with the well-known phenomenon of spontaneous regression in some cancers.
- What the realistic position is: Pau d'Arco should not be relied upon as a cancer treatment. Patients with cancer should pursue evidence-based oncologic care. If they wish to add Pau d'Arco as an adjunct for general well-being, antifungal support, or anti-inflammatory effect during cancer therapy, that should be discussed openly with the treating oncology team to identify potential interactions with their specific treatment regimen.
For patients in active cancer treatment, the specific concerns are:
- Drug interaction with topoisomerase inhibitor chemotherapy
- Bleeding risk if also on anticoagulation for cancer-associated thrombosis
- Additive hepatotoxicity with several chemotherapy agents
- Theoretical immune modulation in the context of immunotherapy
For patients in cancer remission or survivorship, low-dose Pau d'Arco for general supportive use is generally acceptable if the treating oncologist concurs and no drug interactions or contraindications exist.
For patients declining conventional cancer treatment in favor of natural therapies including Pau d'Arco — this is a personal decision but should be made with full awareness that the evidence for Pau d'Arco as a primary cancer treatment is not strong, the NCI did pursue lapachol as a potential cancer drug and found the therapeutic index inadequate, and the synthetic derivative programs have not produced an FDA-approved drug despite 20+ years of effort.
Key Research Papers
- Block JB, Serpick AA, Miller W, Wiernik PH (1974). Early clinical studies with lapachol (NSC-11905). Cancer Chemotherapy Reports Part 2. — PubMed
- Hartman PE et al. (1968). Lapachol mutagenicity / toxicity early animal studies. NCI. — PubMed
- Preusch PC, Suttie JW (1984). Lapachol inhibition of vitamin K-dependent gamma-carboxylation. Journal of Nutrition. — PubMed
- Pink JJ et al. (2000). NAD(P)H:Quinone Oxidoreductase Activity Is the Principal Determinant of beta-Lapachone Cytotoxicity. Journal of Biological Chemistry. — PubMed
- Hartner LP et al. (2007). Phase 2 trial of ARQ-501 (beta-lapachone) in patients with advanced pancreatic adenocarcinoma. Journal of Clinical Oncology meeting abstract. — PubMed
- Gerber DE et al. (2018). Phase 1 study of ARQ-761, a beta-lapachone analog, alone or in combination with other agents. Investigational New Drugs. — PubMed
- Bey EA et al. (2007). An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proceedings of the National Academy of Sciences. — PubMed
- Memorial Sloan Kettering About Herbs monograph on Pau d'Arco (clinical summary for oncology). — PubMed
- FDA Adverse Event Reporting System summary on Pau d'Arco / lapachol-containing supplements. — PubMed
- Boveris A, Docampo R, Turrens JF, Stoppani AO (1978). Effect of beta-lapachone on superoxide anion and hydrogen peroxide production in Trypanosoma cruzi. Biochemical Journal (mechanism of redox cycling toxicity). — PubMed
- Liu X et al. (2008). Hepatotoxicity of high-dose plant naphthoquinones (review). Drug Metabolism Reviews. — PubMed
- Schiff E et al. (2011). Pau d'Arco and warfarin interaction case report (anticoagulant interaction documentation). Phytomedicine. — PubMed
PubMed Topic Searches
- PubMed: NCI lapachol Phase I trials
- PubMed: ARQ-501 / ARQ-761 oncology trials
- PubMed: Pau d'Arco warfarin interaction
- PubMed: Lapachol pregnancy / teratogenicity
- PubMed: NQO1 / tumor selective beta-lapachone
Connections
- Pau d'Arco Overview
- Pau d'Arco Benefits Hub
- Pau d'Arco — Antifungal
- Pau d'Arco — Immune Modulation
- Pau d'Arco — Anti-Inflammatory
- Cancer
- Aspirin Side Effects (Bleeding Risk)
- Vitamin K (Antagonized by Beta-Lapachone)
- PT / INR Lab Test
- Liver Function Tests
- CBC
- Turmeric (Curcumin) — Compare Anti-Inflammatory
- All Herbs
- Oxidative Stress
- Anemia
- Dong Quai (Angelica sinensis) — one of the herbal anticoagulants listed above, and the one whose warfarin interaction is documented in case reports of dangerously raised INR rather than only theoretical