Progressive Supranuclear Palsy
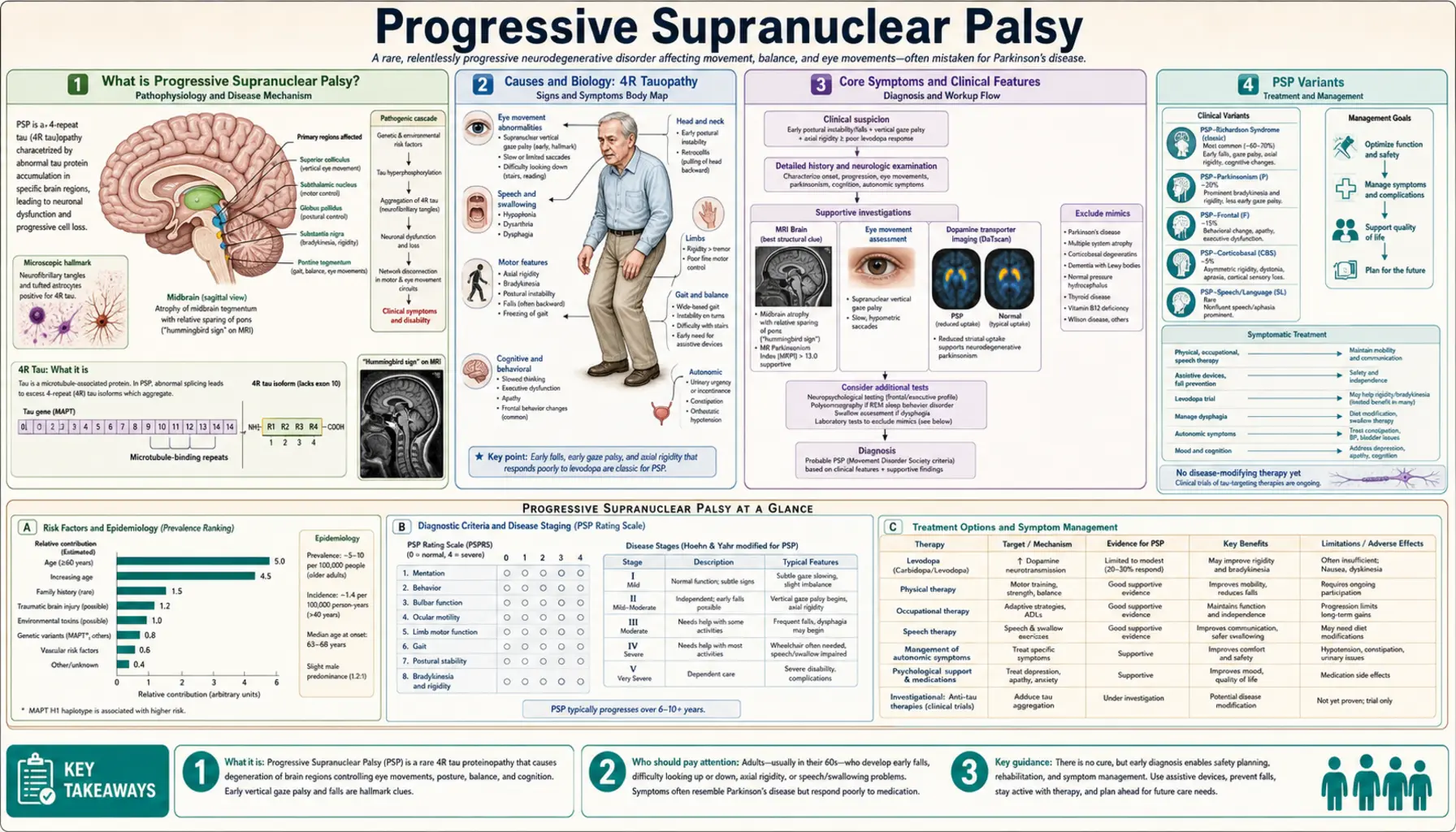
Table of Contents
- What is Progressive Supranuclear Palsy?
- Causes and Biology: 4R Tauopathy
- Core Symptoms and Clinical Features
- PSP Variants
- Diagnosis
- Treatment and Management
- Prognosis and Complications
- Living With PSP
- Research Papers
- Connections
- Featured Videos
What is Progressive Supranuclear Palsy?
Progressive supranuclear palsy (PSP) is a rare, progressive brain disorder and the most common atypical parkinsonian syndrome. It damages nerve cells that control balance, walking, eye movement, speech, and swallowing. The word "supranuclear" refers to where the damage occurs — above (supra) the nerve nuclei that control eye movements.
PSP affects roughly 5–7 people per 100,000. Mean age of onset is around 63 years, and average survival from symptom onset is approximately 7 years — though this varies widely by PSP subtype. It is consistently underdiagnosed, often mistaken for Parkinson's disease or Alzheimer's disease in its early stages.
Causes and Biology: 4R Tauopathy
PSP is classified as a 4R tauopathy. Tau is a protein that normally stabilizes the internal skeleton of neurons. In PSP, tau accumulates abnormally with a specific biochemical pattern — predominantly isoforms containing four microtubule-binding repeat domains (4R tau). This abnormal tau forms tangles inside neurons and glial cells (particularly astrocytes and oligodendrocytes), causing them to malfunction and die.
The midbrain, subthalamic nucleus, globus pallidus, and frontal lobes are most severely affected. Unlike Parkinson's disease, where alpha-synuclein (not tau) is the culprit protein, PSP involves no Lewy bodies.
- Genetic risk: Most cases are sporadic. The MAPT (microtubule-associated protein tau) H1 haplotype on chromosome 17 is a strong risk factor — roughly 95% of PSP patients carry H1/H1 versus ~57% of controls.
- Environmental factors: Exposure to cycad fruit and annonaceous plants (atypical tauopathy clusters in Guadeloupe, Martinique) has been proposed as an environmental trigger, though causality is not established for typical sporadic PSP.
- No inherited mutations: MAPT mutations causing familial tauopathy exist but are exceedingly rare in PSP. The vast majority of patients do not have an identifiable genetic cause.
Core Symptoms and Clinical Features
The classic PSP presentation (Richardson syndrome, PSP-RS) has a characteristic symptom triad that distinguishes it from Parkinson's disease:
1. Early Postural Instability and Falls
Falls in the first year, characteristically backward, are a hallmark of PSP-RS. This stands in sharp contrast to Parkinson's disease, where falls tend to be forward. Patients often report stumbling over nothing or toppling backwards from a standing position. Truncal rigidity (stiffness of the neck and trunk) contributes to this.
2. Supranuclear Vertical Gaze Palsy
Downward gaze palsy is the pathognomonic eye-movement finding. Patients cannot look downward on command, though they can follow moving objects downward if the head is tilted (because the reflex arc bypasses the damaged cortical pathways — this is "supranuclear"). Horizontal gaze is affected later. Patients often show square-wave jerks (involuntary back-and-forth eye movements during attempted fixation) and slow saccades early in the course, before full gaze palsy develops.
Practical consequences: difficulty reading (eyes can't travel down the page), problems with stairs, and difficulty eating (can't look down at food).
3. Dysarthria and Dysphagia
Speech becomes slurred, slow, and low-pitched — a characteristic "growling" quality. Swallowing dysfunction (dysphagia) develops and is a major cause of aspiration pneumonia, the leading cause of death in PSP.
4. Frontal Lobe Dementia
Cognitive changes in PSP are frontal-executive in character: slowed thinking (bradyphrenia), difficulty planning, perseveration, poor insight, and personality changes (apathy, irritability, disinhibition). Memory for recent events is usually relatively preserved compared to Alzheimer's disease.
5. Parkinsonism
Mild parkinsonism is present — axial (neck, trunk) rigidity predominates over limb rigidity. Resting tremor is uncommon. Unlike Parkinson's disease, PSP does not respond well to levodopa.
PSP Variants
PSP is now recognized as a spectrum, not a single uniform syndrome. The Movement Disorder Society (MDS) 2017 criteria define multiple phenotypic variants:
- PSP-RS (Richardson syndrome): Classic presentation. Early falls, vertical gaze palsy, frontal dementia. Most rapid progression, median survival ~5–7 years.
- PSP-P (parkinsonian variant): Resembles Parkinson's disease at onset. Some initial levodopa response (partial, usually lost within 3 years). Falls and gaze palsy emerge later. Median survival longer than PSP-RS (~9 years).
- PSP-PGF (pure akinesia with gait freezing): Prominent gait freezing and speech freezing (pallilalia). Falls occur but gaze palsy and dementia appear late or not at all.
- PSP-CBS (corticobasal syndrome variant): Asymmetric limb apraxia, cortical sensory loss, and alien limb phenomenon — clinically indistinguishable from corticobasal degeneration. PSP pathology at autopsy is found in up to 25% of CBS cases.
- PSP-PLS (primary lateral sclerosis variant): Prominent upper motor neuron signs, pyramidal weakness, and spasticity.
- PSP-F (frontal variant): Behavioral-variant frontotemporal dementia phenotype predominates early. Gaze palsy and falls appear later.
Diagnosis
PSP diagnosis remains primarily clinical, and accuracy improves as the disease progresses.
MRI Imaging
Two classic imaging signs on brain MRI:
- Hummingbird sign (penguin sign): Seen on sagittal midline MRI. Atrophy of the midbrain with a relatively preserved pons creates a silhouette resembling a hummingbird — the midbrain forms the beak. This reflects selective midbrain atrophy characteristic of PSP.
- Morning glory sign: Seen on axial MRI at the level of the superior colliculi. The concave lateral margins of the midbrain tegmentum give a flower-like appearance.
Midbrain-to-pons area ratio below 0.52 and midbrain diameter below 13.5 mm on MRI are diagnostic thresholds validated in published studies.
Tau PET Imaging
Tau PET (using tracers such as flortaucipir/AV-1451 or more recently PI-2620) can detect 4R tau deposition in the basal ganglia, subthalamic nucleus, and dentate nucleus in vivo. It is an important research tool and increasingly used for diagnosis confirmation in ambiguous cases, though it is not yet universally available in clinical practice.
Diagnostic Criteria
The 2017 MDS PSP criteria stratify confidence levels as "probable PSP-RS," "possible PSP-RS," or "suggestive" — requiring combinations of core clinical features (ocular motor, postural instability, akinesia) weighted by how early they appear.
Differentiation from Parkinson's Disease
- PSP: early falls (year 1), backward; PD: falls late in course, forward.
- PSP: downward gaze palsy; PD: gaze relatively preserved.
- PSP: poor levodopa response; PD: robust levodopa response.
- PSP: axial rigidity dominant; PD: appendicular rigidity/tremor dominant.
- PSP: symmetric onset; PD: typically asymmetric onset.
Treatment and Management
There is currently no disease-modifying treatment for PSP. Multiple neuroprotective trials have failed:
- Riluzole (glutamate antagonist): Phase 2 trial showed no benefit on PSP-RS rate of progression.
- Davunetide (microtubule-stabilizing peptide): Phase 2/3 trial failed primary and secondary endpoints.
- Tideglusib (GSK-3β inhibitor, aimed at reducing tau phosphorylation): Phase 2 TAUROS trial showed no significant effect.
- Active research includes antisense oligonucleotides targeting MAPT, tau aggregation inhibitors, and active tau immunotherapy (e.g., gosuranemab, semorinemab — both showed CSF tau lowering but failed clinical endpoints in Phase 2).
Symptomatic Treatment
- Levodopa: A trial is warranted in all patients with parkinsonism — up to 30% achieve modest, temporary benefit (particularly PSP-P variant). Response is partial and usually not sustained beyond 2–3 years. Doses up to 1,200 mg/day may be tried.
- Amantadine: Some patients report modest improvement in bradykinesia or gait freezing.
- Botulinum toxin: For blepharospasm (involuntary eye closure) or cervical dystonia, which can occur in PSP.
- SSRIs/SNRIs: For emotional lability (pathological laughing/crying), depression, or behavioral symptoms.
- Speech and language therapy: Critical for dysphagia management. Modified food textures, thickened liquids, and feeding strategies reduce aspiration risk.
- Physical therapy: Fall prevention strategies, gait aids, and exercise programs to maintain function and reduce injury.
- Occupational therapy: Environmental modification (railings, raised toilet seats, weighted utensils for tremor).
- Gastrostomy (PEG tube): When dysphagia becomes severe and aspiration risk is high, PEG feeding extends survival and prevents aspiration pneumonia, but decision-making requires careful discussion of goals of care.
Prognosis and Complications
PSP is uniformly progressive. Median survival from symptom onset is approximately 7 years for PSP-RS, with a range of 3–15 years. PSP-P and PSP-PGF tend to progress more slowly.
- Aspiration pneumonia is the leading cause of death, resulting from dysphagia and impaired cough reflex.
- Falls and fractures: The combination of backward falls, poor insight, and resistance to using mobility aids results in high rates of serious injury.
- Malnutrition: Dysphagia, cognitive impairment, and motor difficulties with eating lead to weight loss.
- Urinary incontinence develops in most patients as the disease advances.
- Pain: Neck dystonia and rigidity commonly cause neck and shoulder pain.
Living With PSP
PSP is a devastating diagnosis that demands early and honest communication with patients and families. The disease affects the patient's ability to express themselves while insight may initially be preserved, making it especially isolating.
- Fall prevention is urgent from day one. Remove rugs and clutter, install grab bars, and consider a weighted walker (heavier walkers reduce backward tipping) — though many PSP patients resist using aids due to frontal lobe changes affecting insight.
- Eye care: Lubricating eye drops for reduced blink rate, prism glasses for diplopia, and visor caps to shade eyes from glare are practical aids. Reading glasses with prisms to enable downward gaze are sometimes helpful.
- Driving cessation is necessary early — downward gaze palsy and slowed reaction time make driving unsafe.
- Advance care planning should happen early, while the patient can still communicate preferences about PEG tube placement, hospitalization, and end-of-life care.
- Caregiver support: PSP Society (UK) and CurePSP (USA) offer caregiver resources, disease guides, and peer support networks. Caregiver burden is high given the physical demands and behavioral changes.
Research Papers
Curated PubMed topic searches on Progressive Supranuclear Palsy. Each link opens a live PubMed query.
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
Connections
- Neurology
- Parkinson's Disease
- Multiple System Atrophy
- Alzheimer's Disease
- ALS
- Essential Tremor
- Huntington's Disease
- Peripheral Neuropathy
- Multiple Sclerosis
- Depression
- Lion's Mane Mushroom
- Vitamin B12
- Magnesium