G6PD Deficiency (Glucose-6-Phosphate Dehydrogenase Deficiency)
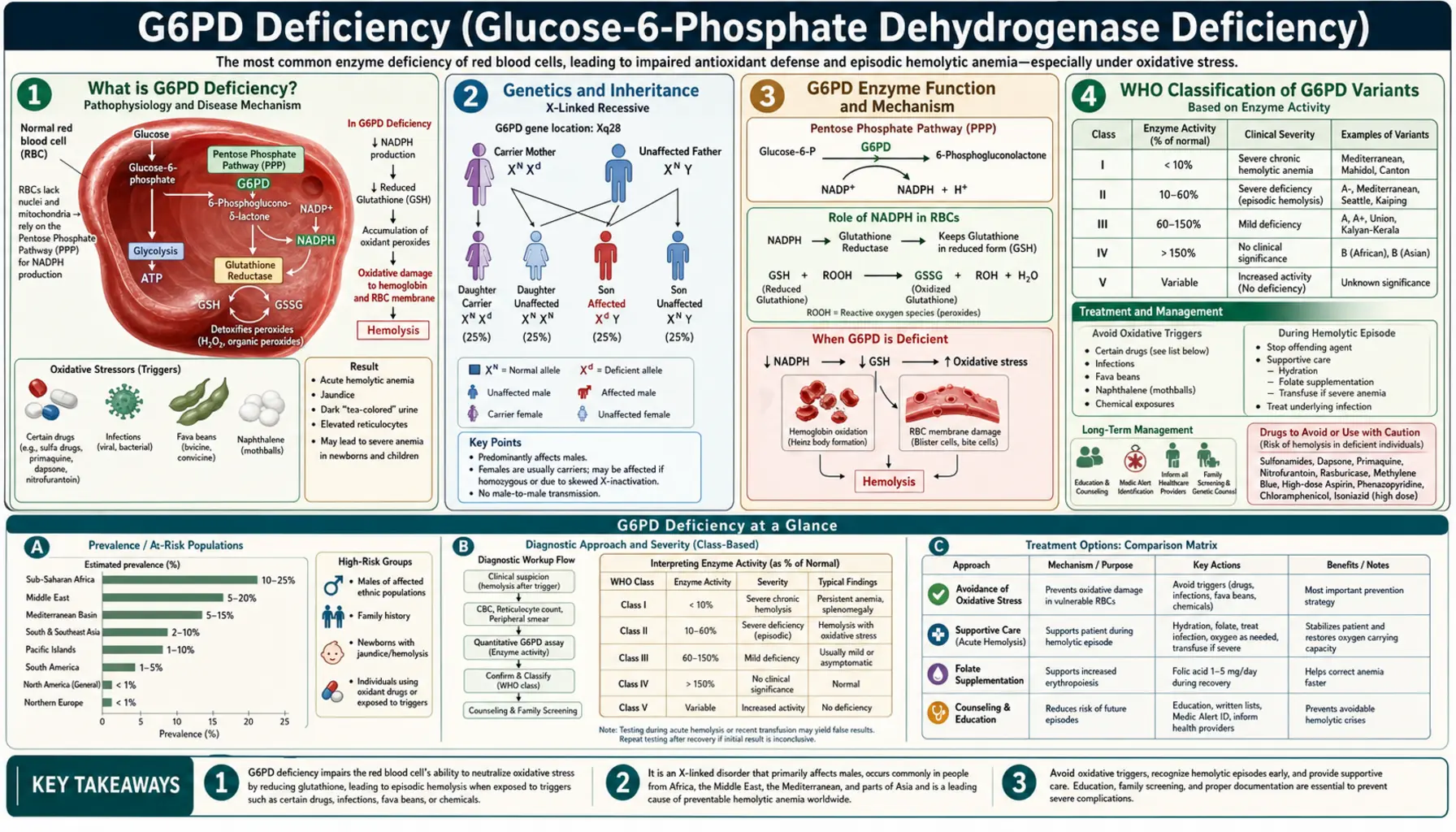
Table of Contents
- What is G6PD Deficiency?
- Genetics and Inheritance
- G6PD Enzyme Function and Mechanism
- WHO Classification of G6PD Variants
- Triggers for Acute Hemolytic Episodes
- Clinical Manifestations
- Diagnosis
- Treatment and Management
- Global Burden and Malaria Protection
- Living with G6PD Deficiency
- Research Papers
- Connections
- Featured Videos
What is G6PD Deficiency?
G6PD deficiency — short for Glucose-6-Phosphate Dehydrogenase deficiency — is the most common enzyme deficiency in the world, affecting an estimated 400 to 500 million people globally. That makes it more common than any other inherited enzyme disorder, more common than sickle cell disease, and roughly comparable in global prevalence to the total number of people with diabetes. Yet most people with G6PD deficiency live their entire lives without knowing they have it, because the condition is almost completely silent between episodes.
The deficiency affects a single enzyme — G6PD — that red blood cells (RBCs) depend on to protect themselves from oxidative damage. When everything is calm, the enzyme's absence causes no problem. But when the body faces an oxidative stress — a serious infection, a particular medication, or even a plate of fava beans — the unprotected red blood cells rupture in a process called hemolysis. The result can range from a mild, self-resolving episode of fatigue and dark urine to a medical emergency requiring blood transfusion.
G6PD deficiency is most prevalent in sub-Saharan Africa, the Mediterranean basin, the Middle East, and Southeast Asia — a distribution that closely mirrors historical malaria transmission zones. This overlap is not coincidental: the same genetic variants that impair RBC oxidative defense also confer partial protection against Plasmodium falciparum, the deadliest malaria parasite. G6PD deficiency is a classic example of a balanced polymorphism, where a genetic disadvantage in one context becomes a survival advantage in another.
Genetics and Inheritance
G6PD deficiency follows an X-linked recessive inheritance pattern. The gene encoding G6PD sits on the long arm of the X chromosome at position Xq28, a densely gene-rich region near the telomere. Because males have only one X chromosome (they are hemizygous), any mutation in their single G6PD gene directly results in deficiency — there is no second copy to compensate. This is why the condition is dramatically more common and more severe in males than in females.
Females carry two X chromosomes and are therefore more likely to be heterozygous carriers — possessing one normal and one deficient copy of the gene. Most heterozygous females have intermediate enzyme activity and remain asymptomatic. However, a fraction of heterozygous females become fully affected through a process called X-inactivation (or lyonization). During early embryonic development, each cell randomly inactivates one of its two X chromosomes. If a female's cells preferentially inactivate the X chromosome carrying the normal G6PD allele — a phenomenon called skewed X-inactivation — her red blood cells will predominantly express the deficient gene, leaving her as clinically vulnerable as an affected male.
Females who are homozygous (carrying two deficient alleles) are as severely affected as males. While this requires inheriting a deficient allele from both parents, it is not rare in high-prevalence populations where consanguineous marriage is common.
The G6PD gene itself spans approximately 18 kilobases and contains 13 exons. More than 230 different point mutations have been identified across human populations, though the vast majority are missense mutations that reduce enzyme activity or stability rather than eliminating it entirely. The clinical consequences depend almost entirely on which mutation is present, since different mutations produce different levels of residual enzyme activity.
G6PD Enzyme Function and Mechanism
To understand why G6PD deficiency causes problems specifically in red blood cells, it helps to understand what the enzyme normally does and why RBCs are uniquely vulnerable without it.
G6PD is the first and rate-limiting enzyme of the pentose phosphate pathway (PPP), also called the hexose monophosphate shunt. In this pathway, glucose-6-phosphate is oxidized, and the energy released is captured not as ATP (as in glycolysis) but as NADPH — a reducing molecule that serves as the cell's primary antioxidant currency.
NADPH's key role in RBCs is to regenerate glutathione (GSH) — the cell's master antioxidant. Glutathione exists in two forms: reduced (GSH, active) and oxidized (GSSG, inactive). The enzyme glutathione reductase uses NADPH to continuously convert GSSG back to GSH, keeping the cell's antioxidant defenses armed. When oxidative stress occurs — from reactive oxygen species generated during infections, drug metabolism, or certain foods — GSH neutralizes those threats before they can damage cellular proteins and lipids.
Here is the critical vulnerability of red blood cells: RBCs have no mitochondria and no nucleus. They cannot carry out aerobic respiration, they cannot synthesize new proteins, and — crucially — they have no alternative source of NADPH. Every other cell type in the body can compensate for low PPP activity using mitochondrial sources of reducing power. RBCs cannot. The pentose phosphate pathway is their only source of NADPH, making G6PD the sole guardian of their oxidative defense.
In G6PD-deficient RBCs exposed to oxidative stress, the cascade of injury proceeds as follows:
- Oxidative stress depletes NADPH faster than G6PD can replenish it.
- Without NADPH, glutathione reductase cannot regenerate GSH; the cell's antioxidant pool collapses.
- Reactive oxygen species oxidize hemoglobin, causing it to denature and precipitate inside the RBC. These precipitates are visible under the microscope as Heinz bodies — named after the physician who first described them, not the condiment.
- Heinz-body-laden RBCs become rigid and are caught in the spleen. Splenic macrophages "bite" off the Heinz body-containing membrane portions, producing bite cells and blister cells on peripheral smear.
- The damaged, rigid RBCs are either removed by the spleen (extravascular hemolysis) or rupture directly in the bloodstream (intravascular hemolysis), releasing hemoglobin into the plasma and causing hemoglobinuria — the characteristic dark urine of a G6PD hemolytic episode.
WHO Classification of G6PD Variants
The World Health Organization developed a five-class system to classify G6PD variants based on enzyme activity and clinical severity. Understanding the class of a patient's variant is clinically important because it predicts their risk for chronic hemolysis versus only episodic hemolysis, and guides decisions about trigger avoidance and monitoring.
Class I — Severe deficiency with chronic nonspherocytic hemolytic anemia (CNSHA): Enzyme activity is less than 10% of normal, and hemolysis is ongoing even without an identifiable trigger. Patients with Class I variants suffer from chronic anemia, persistent jaundice, splenomegaly, and gallstones (from chronic bilirubin overproduction). Class I variants are relatively rare and tend to involve mutations in structurally critical regions of the enzyme. Examples include G6PD Hektoen and G6PD Bangkok.
Class II — Severe deficiency (<10% activity) with episodic hemolysis only: Despite very low enzyme activity, patients with Class II variants are entirely asymptomatic between episodes. The most clinically important Class II variant is G6PD Mediterranean, caused by a c.563C>T mutation that produces a serine-to-phenylalanine substitution at position 188 (S188F). G6PD Mediterranean is the predominant variant across the Mediterranean basin, Middle East, and Indian subcontinent. Patients with this variant are at high risk for severe hemolysis when exposed to fava beans, primaquine, or infection, and they require strict trigger avoidance.
Class III — Moderate deficiency (10–60% activity) with episodic hemolysis: This is the most common class worldwide. The defining example is G6PD A- (also written G6PD A-minus), the predominant variant in people of African descent. G6PD A- is caused by two concurrent mutations: V68M (p.Val68Met) and N126D (p.Asn126Asp). The enzyme produced by G6PD A- has near-normal catalytic activity but is dramatically unstable — it degrades rapidly as red blood cells age. This means that young red blood cells (reticulocytes) in people with G6PD A- have almost normal enzyme activity, while older cells (which circulate for weeks) become severely deficient. This age-dependent vulnerability has an important clinical consequence: when a hemolytic episode is triggered in a G6PD A- patient, the older, deficient RBCs are destroyed, but the younger RBCs survive. The episode is therefore self-limiting, and the patient recovers even without treatment as the bone marrow releases a wave of young, enzyme-replete reticulocytes. This contrasts sharply with G6PD Mediterranean, where even reticulocytes are severely deficient.
Class IV — Normal activity (60–100%): No clinical significance. Includes the normal G6PD B (the wild-type reference) and G6PD A+ (a common polymorphism in Africa with normal activity).
Class V — Increased activity (>150%): Rare variants with supranormal enzyme activity. No established clinical consequence.
Triggers for Acute Hemolytic Episodes
The practical management of G6PD deficiency is largely about identifying and avoiding the triggers that can convert a silent condition into a medical emergency. Triggers are oxidative stressors that overwhelm the already-limited antioxidant capacity of G6PD-deficient red blood cells.
Infections are the single most common trigger for acute hemolytic episodes, accounting for the majority of crises seen in clinical practice. Any significant infection — bacterial pneumonia, viral hepatitis, typhoid fever, influenza — can provoke hemolysis through the massive oxidative burst generated by activated immune cells. Parents of G6PD-deficient children should be aware that a febrile illness may be followed by pallor and dark urine, which are warning signs of hemolysis rather than simply the infection itself.
Antimalarial drugs are historically the most important pharmacological triggers and were instrumental in G6PD deficiency's original discovery during the Korean War, when American soldiers of African descent developed hemolysis after receiving primaquine prophylaxis. Today:
- Primaquine and tafenoquine — both 8-aminoquinoline antiparasitic drugs used to eliminate dormant malaria liver stages (hypnozoites) — are the most potent hemolytic triggers. Tafenoquine is particularly dangerous because its long half-life (15–28 days) means hemolysis cannot be terminated quickly by stopping the drug. G6PD testing is mandatory before prescribing tafenoquine; patients with less than 70% G6PD activity must not receive it.
- Dapsone — used for leprosy, malaria prophylaxis, and Pneumocystis pneumonia prevention — carries significant hemolytic risk.
- Chloroquine and artemisinin derivatives are generally safe at therapeutic doses, though chloroquine can cause mild hemolysis at high doses.
Other medications of concern:
- Cotrimoxazole (trimethoprim-sulfamethoxazole) — the sulfonamide component carries hemolytic risk, particularly in Class II patients.
- Nitrofurantoin — used for urinary tract infections; should be avoided in known G6PD-deficient patients.
- Nalidixic acid — an older fluoroquinolone; modern fluoroquinolones (ciprofloxacin, levofloxacin) are generally considered safe.
- Rasburicase — a uricase enzyme used to prevent tumor lysis syndrome — is an absolute contraindication in G6PD deficiency. Rasburicase generates hydrogen peroxide as a byproduct, causing catastrophic and potentially fatal hemolysis. G6PD testing is required before any rasburicase use.
- Methylene blue — used to treat methemoglobinemia — paradoxically depends on NADPH for its mechanism and is ineffective in G6PD-deficient patients; it may actually worsen hemolysis.
- Aspirin at high doses and phenazopyridine (urinary analgesic) also carry risk.
Fava beans (Vicia faba) — commonly known as broad beans — contain two glycosides, divicine and isouramil, that are potent oxidants directly toxic to G6PD-deficient RBCs. The hemolytic reaction to fava beans is called favism and is one of the oldest recognized food-drug-gene interactions in history, described in ancient Greek texts. Favism is particularly severe in Class II patients (G6PD Mediterranean) and in young children. Even inhalation of fava bean pollen has been reported to trigger episodes. Fava beans are a dietary staple in Mediterranean and Middle Eastern cuisines — their avoidance can be a significant dietary and cultural challenge for affected patients.
Naphthalene — found in traditional moth balls — is a well-documented trigger, particularly relevant for neonates exposed to naphthalene-containing products used in some cultural traditions to store baby clothes. Neonatal hemolysis from naphthalene exposure can be severe.
Clinical Manifestations
The clinical presentation of G6PD deficiency spans a wide spectrum from lifelong complete silence to potentially fatal hemolytic crisis, depending primarily on the patient's WHO variant class and the trigger encountered.
Asymptomatic phase: The overwhelming majority of people with G6PD deficiency are entirely asymptomatic between episodes. They have normal hemoglobin, normal bilirubin, and normal reticulocyte counts. A routine complete blood count gives no hint of the underlying deficiency. This prolonged silence between episodes is both the condition's most reassuring feature and its most dangerous: patients who do not know their diagnosis cannot take precautions.
Acute hemolytic episode: When a trigger is encountered, symptoms typically begin within 24 to 72 hours and include:
- Malaise, fatigue, and pallor from acute anemia as red cells are destroyed faster than they can be replaced.
- Back pain and abdominal pain — often cramping and diffuse — from the systemic hemolytic process.
- Jaundice (yellowing of the skin and sclera) from the bilirubin released as hemoglobin is broken down. Jaundice may appear within hours of onset.
- Dark urine — classically described as "coca-cola colored" or "port wine" urine — from hemoglobinuria (free hemoglobin in the urine). This is a hallmark sign that intravascular hemolysis is occurring.
- Tachycardia and dyspnea in severe cases as the body compensates for falling oxygen-carrying capacity.
In G6PD A- (Class III) patients, the episode is characteristically self-limiting. As older, enzyme-deficient RBCs are destroyed, younger reticulocytes — which have higher enzyme activity — are relatively spared. The bone marrow mounts a reticulocyte response, and the hemoglobin nadir is typically reached by day 7–8, after which the patient recovers spontaneously even without treatment. Hemoglobin rarely falls below 8 g/dL in uncomplicated A- episodes.
In G6PD Mediterranean (Class II) patients, episodes are far more severe because even reticulocytes are enzyme-deficient. Without a protected younger RBC cohort, hemolysis can be catastrophic, with hemoglobin falling precipitously to transfusion-requiring levels.
Neonatal jaundice: G6PD deficiency is one of the most important causes of neonatal hyperbilirubinemia worldwide, particularly in Mediterranean, African, and Asian populations. Unlike ABO or Rh hemolytic disease of the newborn, the jaundice in G6PD deficiency is not purely hemolytic — it also reflects impaired hepatic bilirubin conjugation (neonatal livers are already immature at conjugating bilirubin, and G6PD deficiency further compromises this capacity). Neonatal G6PD-related jaundice can progress to kernicterus — bilirubin deposition in the brain causing permanent neurological damage — if not treated promptly with phototherapy or exchange transfusion. In populations where G6PD deficiency is prevalent, universal newborn screening for jaundice is a public health imperative.
Chronic nonspherocytic hemolytic anemia (CNSHA): Restricted to Class I variants, CNSHA is a state of ongoing hemolysis requiring chronic management: regular monitoring, folate supplementation, transfusion support during crises, and sometimes splenectomy. Gallstones develop early from chronic bilirubin overproduction and may require cholecystectomy.
Diagnosis
Accurate diagnosis of G6PD deficiency requires understanding both the appropriate tests and their important limitations — particularly the critical pitfall of testing during an acute hemolytic episode.
Fluorescent spot test (FST) — screening: The most widely used screening test for G6PD deficiency. A blood spot on filter paper is incubated with glucose-6-phosphate and NADP+; G6PD-sufficient samples produce NADPH, which fluoresces under UV light, while G6PD-deficient samples do not. The FST is inexpensive, rapid, and reliable for identifying hemizygous males and homozygous females. However, it has poor sensitivity for heterozygous females with intermediate activity levels and may produce false normal results in these individuals.
Quantitative G6PD assay — confirmatory: The gold standard for diagnosis is a spectrophotometric measurement of G6PD enzymatic activity expressed as units per gram of hemoglobin (U/gHb). The normal reference range is approximately 4.6–13.5 U/gHb. Class I variants typically show <1 U/gHb; Class II, 1–3 U/gHb; Class III, 3–8 U/gHb. The test is reported relative to hemoglobin to correct for the number of red cells in the sample.
The acute episode pitfall — a critical diagnostic trap: Testing G6PD activity during or immediately after a hemolytic episode routinely yields falsely normal results in G6PD A- patients. The mechanism is straightforward: the oldest, most G6PD-deficient RBCs are selectively destroyed during hemolysis, leaving behind a population enriched in younger reticulocytes with relatively higher enzyme activity. A blood sample taken at the hemoglobin nadir may show enzyme activity in the low-normal range even in a clearly G6PD-deficient patient. For this reason, confirmatory quantitative G6PD testing should be deferred 2–3 months after an acute episode, long enough for the reticulocyte response to settle and the full population of aging, enzyme-deficient RBCs to re-accumulate. This pitfall is less relevant for G6PD Mediterranean patients, whose reticulocytes are also enzyme-deficient, but it has caused missed diagnoses in countless G6PD A- patients in emergency settings.
Peripheral blood smear findings: During an acute episode, a skilled hematology review of the peripheral smear reveals characteristic changes:
- Heinz bodies — visible only with special supravital stains (crystal violet or brilliant cresyl blue), not on a standard Wright-Giemsa smear. They appear as dark purple inclusions abutting the RBC membrane.
- Bite cells (degmacytes) — RBCs that appear to have a "bite" taken out of them, created when splenic macrophages remove Heinz body-laden membrane segments.
- Blister cells — RBCs with an apparently empty vacuole under the membrane where Heinz bodies have been removed.
- Polychromasia — a bluish tint to some RBCs reflecting reticulocytosis (the bone marrow's compensatory response).
Newborn screening: Many countries with high G6PD prevalence, including Greece, Cyprus, Hong Kong, Singapore, and Saudi Arabia, incorporate G6PD deficiency into their universal newborn screening programs. In the United States, newborn screening for G6PD is not universal but is offered in some states.
Treatment and Management
There is no cure for G6PD deficiency, and most patients never require medical treatment. Management centers on education, trigger avoidance, and prompt response to acute episodes when they occur.
Patient education — the cornerstone of management: Every patient diagnosed with G6PD deficiency should receive comprehensive education covering:
- The specific foods to avoid (fava beans above all; some sources also list blueberries, soya products, and tonic water [contains quinine] as lower-level concerns, though fava beans are the only clearly documented dietary trigger in clinical practice).
- A complete medication list of drugs to avoid or use with caution.
- Warning signs of a hemolytic episode (dark urine, jaundice, rapid fatigue) and when to seek emergency care.
- The importance of informing all healthcare providers and pharmacists of their diagnosis before any new medication is prescribed.
Medical alert identification: Patients with Class I or Class II variants should wear a medical alert bracelet or carry a medical alert card specifying their diagnosis and the absolute contraindications (rasburicase, tafenoquine, methylene blue). In an emergency, this information can prevent inadvertent administration of a triggering drug.
Management of acute hemolytic episodes:
- Remove the trigger: Stop the offending drug immediately. Treat the precipitating infection if present.
- Hydration: Intravenous fluids to maintain urine output and protect the kidneys from hemoglobin precipitation in the tubules (which can cause acute kidney injury).
- Blood transfusion: Indicated when hemoglobin falls below approximately 7 g/dL or when the fall is rapid regardless of absolute level, or when the patient has cardiovascular compromise. Packed RBCs from a G6PD-normal donor provide functional cells unaffected by the trigger.
- Folate supplementation: During a hemolytic episode, the bone marrow's accelerated erythropoiesis can deplete folate stores. Supplementation with 1–5 mg folic acid daily supports recovery.
- Phototherapy or exchange transfusion for neonatal hyperbilirubinemia, following standard thresholds based on gestational age and bilirubin level.
Primaquine dosing strategy in G6PD A- patients: Radical cure of vivax or ovale malaria requires elimination of dormant liver stages (hypnozoites), which requires primaquine. For Class III (G6PD A-) patients, a supervised weekly high-dose primaquine regimen (0.75 mg/kg weekly for 8 weeks) is an alternative to the standard daily regimen, taking advantage of the self-limiting hemolysis in A- patients while allowing bone marrow recovery between doses. This strategy is not appropriate for Class I or II patients.
Management of CNSHA (Class I variants): These patients require hematology specialist care. Splenectomy may reduce transfusion requirements by removing the primary site of extravascular hemolysis. Folate supplementation is ongoing. Avoidance of all trigger medications and foods is mandatory, as even a small additional oxidative burden can precipitate a crisis in an already-hemolyzed patient.
Emerging therapies: Gene therapy trials targeting G6PD deficiency are in early-stage development, offering the prospect of a one-time curative treatment for Class I CNSHA patients. Small-molecule pharmacological chaperones that stabilize mutant G6PD protein are also under investigation.
Global Burden and Malaria Protection
G6PD deficiency is distributed across the globe in a pattern that reflects ancient and ongoing selection pressure from malaria. The highest allele frequencies — often 15–25% of males in affected populations — are found in sub-Saharan Africa, the Mediterranean basin, the Middle East, the Indian subcontinent, and Southeast Asia. In some hyperendemic malaria regions, such as parts of West Africa and Papua New Guinea, G6PD deficiency allele frequencies reach 30% or higher.
The malaria-protective mechanism of G6PD deficiency has been studied for decades and is now reasonably well characterized. G6PD-deficient RBCs appear to resist Plasmodium falciparum infection through multiple mechanisms:
- The parasite requires NADPH for its own metabolism; reduced NADPH availability in deficient cells impairs parasite growth.
- G6PD-deficient RBCs expressing ring-stage parasites are removed from circulation by the spleen more rapidly than normal infected cells, limiting parasite multiplication.
- The oxidative environment inside deficient cells may directly damage the parasite.
Epidemiological studies have estimated that G6PD deficiency provides approximately 46–58% protection against severe malaria in heterozygous females and a similar degree of protection in hemizygous males (though males experience the tradeoff of greater hemolytic risk). This protection rivals the protection conferred by sickle cell trait (HbAS), making G6PD deficiency one of the most powerful malaria-resistance polymorphisms in the human genome.
This protective relationship has major implications for malaria eradication programs. The most effective drugs for clearing malaria liver stages — primaquine and tafenoquine — are precisely the drugs most dangerous to G6PD-deficient individuals. Mass drug administration programs using these agents must incorporate G6PD testing to avoid triggering hemolytic crises at population scale. The development of rapid, field-deployable G6PD diagnostic tests has become a public health priority for the WHO's malaria elimination agenda.
Living with G6PD Deficiency
For the vast majority of people with G6PD deficiency, the condition is compatible with a completely normal life, with one important caveat: they must be informed and their healthcare providers must be aware. The key practical steps are straightforward but non-negotiable.
Know your variant class. If you have been diagnosed with G6PD deficiency, ask your doctor which class your variant belongs to. Class III (G6PD A-) patients have a much more forgiving margin than Class I or II patients, but all classes require trigger avoidance.
Carry your diagnosis with you. In an emergency room or surgical setting, a provider who doesn't know your diagnosis may administer rasburicase (for hyperuricemia from tumor lysis) or prescribe methylene blue (for methemoglobinemia) — both potentially catastrophic in G6PD deficiency. A medical alert bracelet or a note in your emergency contact information can prevent this.
Fava beans are not negotiable. In Mediterranean and Middle Eastern households where ful medames, bessara, or other fava-bean dishes are cultural staples, avoidance can be socially difficult. But favism episodes can be severe — even fatal in young children — and no tolerance-building or desensitization exists. This is one dietary restriction that must be followed strictly.
Children and schools. Parents of G6PD-deficient children should inform school nurses and teachers, particularly about fava bean avoidance (many school lunch programs in Mediterranean-origin communities serve them) and about the risk of naphthalene exposure from mothball-stored clothing.
Pregnancy. G6PD-deficient mothers carrying G6PD-deficient fetuses may experience fetal or neonatal hemolysis. Neonatologists should be informed when a G6PD-deficient mother delivers, so the neonate can be monitored for jaundice and treated promptly.
Research Papers
- Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis. 2009;42(3):267–278. — Search PubMed
- Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371(9606):64–74. — Search PubMed
- Luzzatto L, Mehta A, Vulliamy T. Glucose-6-phosphate dehydrogenase deficiency. In: Scriver CR et al., eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill; 2001. — Search PubMed
- Howes RE, Piel FB, Patil AP, et al. G6PD deficiency prevalence and estimates of affected populations in malaria endemic countries: a geostatistical model-based map. PLoS Med. 2012;9(11):e1001339. PMID 23152723
- Youngster I, Arcavi L, Schechmaster R, et al. Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf. 2010;33(9):713–726. — Search PubMed
- Beutler E. G6PD deficiency. Blood. 1994;84(11):3613–3636. PMID 7949118
- WHO Working Group. Glucose-6-phosphate dehydrogenase deficiency. Bull World Health Organ. 1989;67(6):601–611. PMID 2633878
- Ruwende C, Khoo SC, Snow RW, et al. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 1995;376(6537):246–249. PMID 7617034
- Luzzatto L, Ally M, Notaro R. Glucose-6-phosphate dehydrogenase deficiency. Haematologica. 2020;105(12):2753–2759. — Search PubMed
- Minucci A, Moradkhani K, Hwang MJ, et al. Glucose-6-phosphate dehydrogenase (G6PD) mutations database: review of the "old" and update of the new mutations. Blood Cells Mol Dis. 2012;48(3):154–165. — Search PubMed
- Belfield KD, Tichy EM. Review and drug therapy implications of glucose-6-phosphate dehydrogenase deficiency. Am J Health Syst Pharm. 2018;75(3):97–104. — Search PubMed
- Recht J, Ashley EA, White NJ. Safety of 8-aminoquinoline antimalarial medicines. WHO Evidence Review Paper. 2014. — Search PubMed
Connections
- Hematology
- Anemia
- Hemolytic-Uremic Syndrome
- Hereditary Spherocytosis
- Thalassemia
- Paroxysmal Nocturnal Hemoglobinuria
- Sickle Cell Disease
- Aplastic Anemia
- Thrombocytopenia
- Complete Blood Count
- Iron
- Folate (B9)
- Jaundice
- Malaria — G6PD variants confer partial resistance; antimalarials trigger hemolysis.